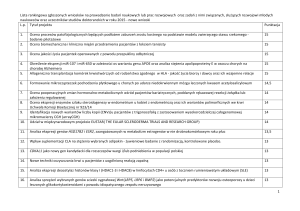
ALEKSANDRA ŚWIERCZ
Co to jest transkryptom?
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
2
Ekspresja genów
http://genome.wellcome.ac.uk/doc_WTD020757.html
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
3
Co to jest mikromacierz?
Mikromacierz DNA (określany także jako chip DNA) to zbiór, krótkich DNA
przyczepionych do powierzchni szklanej płytki.
Mikromacierzy można użyć do mierzenia poziomu ekspresji genów
Każdy punkt na mikromacierzy zawiera specyficzną sekwencję DNA, która
reprezentuje jeden z genów (sonda, ang. probe)
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
5
Macierz ekspresji genów
Próbki
Próbka 1 Próbka 2 Próbka 3 Próbka 4 Próbka 5
Geny/
sondy
…
1
0.25
0.30
0.70
1.53
0.90
…
2
-0.12
0.30
0.45
0.12
0.40
…
3
0.13
0.46
0.75
0.32
0.53
…
4
-0.16
-0.43
-0.65
-0.79
-0.32
…
...
…
…
…
…
…
…
Poziom ekspresji genu lub stosunek, dla genu i-tego
w j-tej próbce mRNA
M=
A=
A. Świercz
{
{
log2(red intensity/green intensity)
Funkcja (PM,MM) MAS, dchip lub RMA
½ log2(red intensity*green intensity)
Funkcja (PM,MM) MAS, dchip lub RMA
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
6
Różnice między eksperymentem
mikromacierzowym a RNA-seq
Przy użyciu mikromacierzy można badać poziom ekspresji znanych
genów, natomiast wykorzystując RNA-seq można także wykryć nowe
izoformy genów
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
7
RNA-seq razem z Ion Torent
http://www.youtube.com/watch?v=V_4n8n5Z6I8
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
8
Dlaczego RNA-seq zamiast DNA-seq?
Badanie funkcjonalności
◦ Genom może być taki sam, ale warunki eksperymentalne mogą mieć wpływ na
ekspresję genów (np. traktowanie komórek lekarstwem, vs niczym nietraktowane,
lub mysz dzika vs zmieniona genetycznie)
Niektóre zmiany mogą być widoczne dopiero na poziome RNA
◦ Alternatywne izoformy
◦ Fuzja transkryptów (trans-splicing, transcription-induced chimerism)
◦ Edytowanie RNA - zmiana informacji w transkrypcie RNA przez reakcję chemiczną
powodującą zmianę jednej zasady azotowej w inną (C->U, A->I, Inozyna
interpretowana jako G).
Przewidywanie sekwencji transkryptów z sekwencji genomu jest trudne:
◦ Alternatywny transkrypt
◦ Edytowanie RNA
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
9
Dlaczego RNA-seq zamiast DNA-seq?
Interpretacja, czy poszczególne mutacje mają wpływ na sekwencje
białkową
◦ Mutacje ‚regulujące’ które wpływają na to czy izoformy mRNA ulegają
ekspresji i jak dużej
◦ Czy mutacje wpływają na promotory, eksonowe/intronowe motywy, miejsca
splicingowe?
Wpływ na białka kodujące mutacje somatyczne (często
heterozygotyczne)
◦ Jeśli gen nie ulega ekspresji, mutacja w takim genie będzie mniej interesująca
◦ Jeśli gen ulega ekspresji tylko z alleli dzikiego typu, może to sugerować na
utratę funkcjonalności (haploinsufficiency)
◦ Jeśli allel mutanta ulega ekspresji, może to oznaczać kandydata na target dla
leku
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
10
Do czego wykorzystywane jest RNA-seq?
Badanie ekspresji genów oraz różnicowej ekspresji genów
Wyszukiwanie alternatywnego splicingu w genach
Odkrywanie nowych transkryptów/izoform
Odkrywanie mutacji w genach
Wykrywanie fuzji genów
Edytowanie RNA (mutacje w RNA)
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
11
Mikromacierze vs sekwencjonowanie
Porównanie eksperymentów mikromacierzowych i RNA-seq pokazało, że:
◦ Jest duża zgodność w wynikach pomiędzy platformami, w szczególności
pomiędzy wykrywaniem różnicowej ekspresji genów
◦ Platforma sekwencjonowania jest bardziej wrażliwa na wykrycie zmian, jest
bardziej odporna na tło i różnice w powtórzeniach technicznych
◦ Zaletą RNA-seq jest porównanie poziomu ekspresji różnych genów między sobą
(dla mikromacierzy można porównać ten sam gen między różnymi warunkami)
◦ Ograniczeniem RNA-seq jest natomiast wykrzywienie GC oraz
niejednoznaczność w mapowaniu
◦ Większa jest moc statystyczna w wykrywaniu zmian, gdy odczyty występują w
większej liczności
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
12
Sekwencjonowanie RNA – po kolei
RNA-seq Module, 2013, www.bioinformatics.ca
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
14
Trzy podejścia do mapowania RNA-seq
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
15
Trudności przy mapowaniu RNA
Geny w genomach eukariotycznych zawierają introny, a sewkencje
mRNA są już ich pozbawione. Programy mapujące odczyty z
eksperymentów RNA-seq muszą być w stanie dopasować sekwencje z
przerwami
◦
◦
◦
◦
Introny w genomach ssaków mają długość od 50 bp - 100,000 bp.
Średnia długość transkryptu mRNA u człowieka to 2227 bp
Średnia długość eksonu to 235 bp
Średnio w jednym genie jest 9 eksonów
Około 20% odczytów które mapują się na łączeniach eksonów mapują
się tylko na < 10 nukleotydach na drugim eksonie
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
16
Trudności przy mapowaniu RNA
Część sekwencji pochodzi z przetworzonych pseudogenów, z których
niektóre lub wszystkie introny zostały usunięte (może to spowodować
nieprawidłowe mapowanie odczytów)
◦ Genom ludzki posiada 14tys pseudogenów
◦ Pseudogeny mają sekwencję bardzo podobną do funkcjonalnych genów
zawierających introny. W większości przypadków nie ulegają transkrypcji
◦ Problem w mapowaniu wynika stąd że odczyty, które mapują się na łączeniu
eksonów, będą się mapowały w całości dokładnie lub z niewielkim błędem
do pseudogenów, które nie zawierają intronów.
◦ Jeśli metoda mapująca mapuje najpierw odczyty w całości, a resztę próbuje
dopasować z podziałem na eksony, to pominie odczyty które w całości
zmapowane zostały do pseudogenów
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
17
D. Kim, G. Pertea, C. Trapnell, H. Pimentel, R. Kelley, S.L. Salzberg „TopHat2: accurate alignment of
transcriptomes in the presence of insertions, deletions and gene fusions” Genome Biology 2013, 14:R36
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
18
Trudności przy mapowaniu RNA
Transkrypt badanego genomu może się różnić od genomu
referencyjnego
◦ Różnice mogą być małe, typu SNP, insercje, delecje, niedopasowania
◦ Zmiany mogą być większe, rearanżacje chromosomowe – przeniesienia
dłuższych fragmentów, wiele kopii
◦ Małe zmiany nie wpływają znacznie na mapowanie – trzeba dopuścić
możliwość błędów w niedopasowaniu (może to niestety spowodować wiele
miejsc mapowania)
◦ Większe zmiany: duże usunięcia, inwersje w obrębie tego samego
chromosomu, oraz translokacje między-chromosomowe powodują że trudno
znaleźć kolejne eksony genu
fragment chrom. 2
fragment chrom. 5
W genomie badanym w stosunku do genomu referencyjnego
część genu uległa translokacji oraz inwersji
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
19
TopHat2 pipeline
Znane sygnały podziału eksonów
GT-AG, GC-AG, AT-AC
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
20
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
21
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
22
Alamancos GP, Agirre E,
Eyras E. (2014)
Methods to study splicing
from high-throughput
RNA sequencing data.
Methods Mol Biol
1126:357-97.
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
24