ALEKSANDRA ŚWIERCZ
Ekspresja genów
http://genome.wellcome.ac.uk/doc_WTD020757.html
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
2
Różnice między eksperymentem
mikromacierzowym a RNA-seq
Przy użyciu mikromacierzy można badać poziom ekspresji znanych
genów, natomiast wykorzystując RNA-seq można także wykryć nowe
izoformy genów
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
3
Do czego wykorzystywane jest RNA-seq?
Badanie ekspresji genów oraz różnicowej ekspresji genów
Wyszukiwanie alternatywnego splicingu w genach
Odkrywanie nowych transkryptów/izoform
Odkrywanie mutacji w genach
Wykrywanie fuzji genów
Edytowanie RNA (mutacje w RNA)
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
4
Sekwencjonowanie RNA – po kolei
RNA-seq Module, 2013, www.bioinformatics.ca
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
5
Trzy podejścia do mapowania RNA-seq
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
6
Mapowanie – wyznaczenie
poziomu ekspresji – wizualizacja
TopHat / BowTie
Cufflinks
Cuffmerge
Cuffdiff
Cummerbund
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
11
TopHat2 pipeline
Znane sygnały podziału
GT-AG, GC-AG, AT-AC
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
12
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
13
Jak wyrażana jest ekspresja genu?
RPKM – Reads Per Kilobase of transcript per Million mapped reads
FPKM – Fragments Per Kilobase of transcript per Million mapped reads
W RNA-Seq – poziom ekspresji transkryptu jest proporcjonalny do liczby
fragmentów cDNA z którego pochodzi. Chociaż:
◦ Liczba fragmentów jest przechylona w kierunku większych genów
◦ Całkowita liczba fragmentów jest uzależniona od głębokości sekwencjonowania
RPKM (FPKM) = (109 * C) / (N * L)
◦ C – liczba zmapowanych odczytów (fragmentów) do genu/transkryptu/eksonu
◦ N – całkowita liczba zmapowanych odczytów (fragmentów) w bibliotece
◦ L – liczba nukleotydów w genie/transkrypcie/eksonie
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
14
Alternatywny splicing
W procesie splicingu łączone są ze sobą różne eksony z pre-mRNA na różne
sposoby, czasami z pominięciem niektórych eksonów, lub z zachowaniem
niektórych intronów
Jeśli warianty splicingowe dotyczą sekwencji kodującej, powstałe białka
różnią się sekwencją aminokwasową, co może powodować np.
zróżnicowanie funkcji.
Jeśli warianty splicingowe dotyczą obszarów niekodujących może to
wpływać np. na wzmocnienie translacji lub stabilność mRNA.
Rekordem w liczbie różnych wariantów splicingowych jest gen Dscam D.
melanogaster, który ma ponad 38 tys. różnych wariantów (więcej niż liczba
wszystkich genów) *
* C.
Ghigna, C. Valacca, G. Biamonti „Alternative Splicing and Tumor Progression”, Curr
Genomics. Dec 2008; 9(8): 556–570.
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
16
Różne
warianty
splicingowe
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
17
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
18
Różne warianty splicingowe
Mutually exclusive exons
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
19
Zhiyong Guo, Yun Qiu
A New Trick of an Old
Molecule: Androgen
Receptor Splice
Variants Taking the
Stage?!
Int J Biol Sci 2011;
7(6):815-822.
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
20
Jak Cufflinks radzi
sobie z wykrywaniem
alternatywnego
splicingu?
C. Trapnell, BA Williams, G Pertea, A Mortazavi, G Kwan, MJ
van Baren, SL Salzberg, BJ Wold, L Pachter, „Transcript
assembly and quantification by RNA-Seq reveals
unannotated transcripts and isoform switching during cell
differentiation, Nature Biotechnology 28(5) 2010, p. 511515
C Trapnell, DG Hendrickson, M Sauvageau, L Goff, JL Rinn, L
Pachter, „Differential analysis of gene regulation at
transcript resolution with RNA-seq”, Nature Biotechnology
31(1), 2013, p. 46-53.
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
21
Mapowanie odczytów sparowanych za pomocą TopHat-a.
Każda para odczytów traktowana jest jako jedno
dopasowanie. Odczyty mogą być zmapowane w całości,
lub z podziałem pomiędzy eksonami
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
22
Cufflinks
W pierwszym kroku wyszukiwane są pary
„niekompatybilnych” fragmentów, które muszą
pochodzić z innych izoform mRNA (zaznaczone na
żółto, niebiesko i czerwono).
Fragmenty (sparowane odczyty), są wierzchołkami w
grafie. Wierzchołki są łączone pomiędzy parami
kompatybilnych fragmentów.
Szarym kolorem zaznaczone są fragmenty, które
mogą pochodzić z dowolnych transkryptów.
Ścieżki w grafie odpowiadają wzajemnie
wykluczającym się fragmentom, które mogą być
połączone w izoformy.
Graf może być pokryty minimalnie przez 3 ścieżki –
oznaczone 3 kolorami, co w efekcie daje 3 odrębne
izoformy
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
23
Cufflinks
Fragmenty są znakowane (tutaj kolorem) w zależności
od tego z której izoformy pochodzą. Fioletowy fragment
może pochodzić z niebieskiego lub czerwonego. Szare
fragmenty mogą pochodzić z dowolnej izoformy.
Cufflinks estymuje liczność transkryptu używając
modelu statystycznego, w którym prawdopodobieństwo
obserwowania każdego fragmentu jest liniową funkcją
liczności transkryptów, z których mogą pochodzić.
Ponieważ długość sekwencjonowanych fragmentów nie
jest znana (sparowane odczyty są końcami
fragmentów), a przypisanie fragmentu do różnych
izoform powoduje że różna jest jego długość – Cufflinks
wyznacza rozkład długości odczytów. Rozkład ten jest
następnie wykorzystywany do przypisania fragmentów
do różnych izoform (fioletowy fragment byłby zbyt
długi, gdyby został przypisany do czerwonego
transkryptu).
W ostatnim kroku program maksymalizuje prawdopodobieństwo liczności każdej z
izoform i przydziela im odpowiednio numeryczne wartości (γ1, γ2, γ3)
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
24
Cufflinks
W powyższym przykładzie analizowany był tylko fragment jednego genu. Wszystkie
fragmenty genu należy potem skleić całość
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
25
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
26
Do czego jest potrzebny cuffmerge?
Pozwala na łączenie wyników z działania cufflinks’a dla różnych próbek
◦ Jest to potrzebne ponieważ dla każdej próbki cufflinks może wykryć inną
liczbę oraz inną strukturę transkryptów
Odfiltrowywane są transkrypty, które są najprawdopodobniej
artefaktami (transfrags)
Opcjonalnie może także podać plik GTF w odniesieniu do genomu
referencyjnego, w którym połączone będą dotychczas znane oraz nowe
izoformy wraz z maksymalizacją jakości zasemblowanych transkryptów
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
27
Jak wyznaczyć ekspresję transkryptu?
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
28
Jak działa cuffdiff?
• Modelowanie zmienności w liczbie fragmentów dla każdego
genu dla różnych powtórzeń – estymacja wariacji
• Liczba fragmentów dla każdej izoformy jest estymowana dla
każdego powtórzenia (jak poprzednio) razem z miarą
niepewności pochodzącą od niejednoznacznie
zmapowanych odczytów
• Transkrypty, z większą liczbą współdzielonych eksonów, a
niewielką liczbą jednoznacznie przypisanych fragmentów będą
miały mniejszą niepewność
• Algorytm łączy estymowaną niepewność razem ze
zmiennością pomiędzy powtórzeniami poprzez model
ujemnego rozkładu dwumianowego dla liczby fragmentów,
w celu estymowania liczby niezgodności dla każdego
transkryptu w każdej bibliotece
• Te estymowane niezgodności używane podczas testowania
statystycznego pozwalają na znalezienie znaczących
statystycznie genów i transkryptów, które uległy
zróżnicowanej ekspresji
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
29
Wizualizacja, np. cummeRbund
Automatycznie generuje zestaw wykresów do porównania ekspresji dla
różnych (zadanych) próbek
◦
◦
◦
◦
◦
Wykresy z rozkładem wartości
Wykresy z korelacją
Wykresy MA
Wykresy ‚volcano’
Wykresy klastrowania, PCA, MDS – w celu ogólnej oceny związku pomiędzy
warunkami
◦ Heatmapy – wykresy gęstości
◦ Wykresy z poziomu genów lub transkryptów pokazujące strukturę
transkryptów i poziom ekspresji
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
30
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
31
`
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
32
Alternatywne do FPKM/RPKM
„Raw counts” – liczba odczytów/fragmentów przypadająca na
gen/transkrypt
„HTSeq” – htseq-count – zlicza liczbę odczytów przypadających na
gen/ekson
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
33
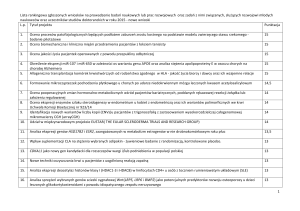
FPKM czy ‚raw’ counts ?
FPKM
◦ Gdy chcemy wykorzystać zalety ‚smokingu’ (tophat/cufflinks/itp)
◦ Można wykorzystać do wizualizacji na heatmapie
◦ Także do wyznaczania zmiany poziomu ekspresji genów
Surowa liczba odczytów
◦ Jeśli chcemy wykorzystać bardziej zaawansowane metody statystyczne do
normalizacji, czy do badania zmiany poziomu ekspresji genów
◦ Do wykorzystania przy skomplikowanych projektach eksperymentalnych, przy
analizie trendów czasowych i przy wykorzystaniu innych zaawansowanych
testów statystycznych
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
34
Limma
Liniowy model początkowo używany do analizy danych pochodzących z
eksperymentów mikromacierzowych, następnie przystosowany do
analizy danych RNA-seq
Umożliwia na bardzo rozbudowany model porównania, poprzez
zdefiniowanie design matrix oraz contrast matrix.
> design <- model.matrix(~ 0+factor(c(1,1,1,2,2,3,3,3)))
> colnames(design) <- c("group1", "group2", "group3")
> contrast.matrix <- makeContrasts(group2-group1, group3group2, group3-group1, levels=design)
Umożliwia analizę trendów
Time points
1d 2d
4d
8d
12d
Control vs treated
http://www.bioconductor.org/packages/release/bioc/html/limma.html
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
36
DESeq
Utworzony dla danych RNA-seq. Wymaga tabeli z wyznaczoną liczbą
odczytów przypadających na dany gen.
Normalizacja względem liczby wszystkich odczytów przypadających na
próbkę
> cds = estimateSizeFactors( cds )
Estymacja wariancji – wyznaczenie jak bardzo geny różnią się w
ekspresji pomiędzy różnymi próbkami
> cds = estimateDispersions( cds )
Wyznaczenie różnicowej ekspresji
> res = nbinomTest( cds, "untreated", "treated" )
http://www.bioconductor.org/packages/release/bioc/html/DESeq.html
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
37
Różne programy – porównanie
wyników
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
38
Alamancos GP, Agirre E,
Eyras E. (2014)
Methods to study splicing
from high-throughput
RNA sequencing data.
Methods Mol Biol
1126:357-97.
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
39