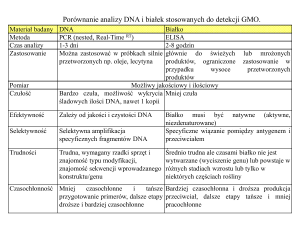
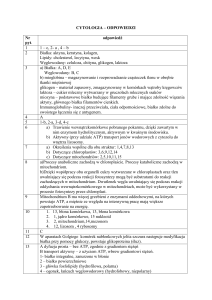
Choroby prionowe
-
zawsze śmiertelne
rzadkie
od momentu zakażenia nawet ponad 20 lat stabilnych itd., potem nagle bum
I objaw – demencja
Dotyczą zawsze mózgu, priony uszkadzają neurony, określenie chorób prionowych –
encephalopatie gąbczaste
Zapadają na nie wszystkie ssaki
Przyczyna – zakażenie lub zmiany genetyczne
Choroba dziedziczna
W tkance jeszcze nie naruszonej występują płytki amyloidu (ważne w diagnostyce,
barwnik cago – czerwony pod wpływem amyloidu -> pomarańczowy, tioflamina
powoduje, że płytki świeca na zielonkawo)
U człowieka występują 2 jednostki chorobowe:
1. Kuru (hist. Na Nowej Gwinei występuje szczep tubylców Fore – kanibale spożywali
mózgi zmarłych, u niektórych występowały demenja, drżenie kończyn,
charakterystyczny grymas uśmiechu na twarzy – stąd choroba śmiejącej się śmierci)
Guy Dusek stwierdził obecność zmian patofizjologicznych w mózgu, doświadczalni wykazał,
że to choroba zakaźna (szczepił szympansy fragmentami mózgu), otrzymał Nobla.
2. Kreutzfelda – Jacoba
Występuje w odmianach:
A) sporadyczna fsCJD
nie ma charakteru zakaźnego, ne udowodniono ze jest genetyczna, mogą chorować
również ludzie młodzi
objawy: postępujące otępienie, ruchy mimowolne, czasem utrata słuchu
B) rodzinna rCJD
dziedziczna, mutacja w genie PrpP.
Jednostki postaci rodzinnej:
2a śmiertelna dziedziczna bezsenność
2 – 3 tygodnie bez snu
2b zespół Gertsmana – Streiflera (na egzaminie pisać: zespół GSS)
opisany w ’36 w Austrii, 2001 – I przypadek w Polsce
narastające otępienie
3 postać jatrogenna jCJD
przenoszona przy niezawinionym udziale lekarza bądź pielęgniarki
1920 – pierwsze przypadki, do zakażenia doszło przy próbach przeszczepienia
rogówki, częstsze przy przeszczepie przysadki (opisanych ponad 100
przypadków), przy przeszczepie opony twardej (do zakażeń dochodzi często
w neurochirurgii),
1 z przyczyn – leczenie gonadotropiną, występuje raczej w młodym wieku
4 Wariant choroby Kreutzfelda – Jacoba vCJD
łączy się ściśle z BSE, pierwsze przypadki opisane w Edynburgu, tylko w
Angli 120 przypadków
objawy: silne pobudzenie, agresja, urojenia, otępienie, zaburzenia równowagi!
U zwierząt:
1) najstarsza choroba prionowa – Scraie
pierwsze doiesienia 1770
silne swędzenie skóry, owce wygryzały sobie sierść, zakaźna biegunka, śmierć po roku –
dwóch, mózgi jak gąbki
2) choroba szalonych krów
uciekają od ludzi, splątany chód
zmielone kości padłych owiec dodawane były do paszy
owca -> bydło -> CJD, ludzie
czynnik etiologiczny:
badania Prusinera (Nobel ’97)
teza: czyste białko ma charakter zakaźny
charakterystyczna część białka
w chromosomie 21 homologiczny odcinek
gen PrpP koduje białko prionowe prpk (komórkowe – fizjologiczne)
u chorych białko prpsc
rola białka polega na regulowaniu poziomu wapnia w neuronie. prpsc powoduje zwiększenie
liczby prionów w neuronach, a w konsekwencji zjawisko programowanej śmierci
*Lampy promieniotwórcze nie działają na priony
Czasem przy chorobach prionowych pojawia się enzym enolaza neuronowa.
Różnice między Prpk i Prpsc
- podatność na enzymy proteolityczne – 2. niewrażliwe
- w budowie. Białko 1. może przekształcać się 2. zbiera się go dużo, neurony pękają,
zakażają się kolejne (powoli)