ISBN 978-83-62110-23-0
EGZEMPLARZ BEZPŁATNY
CZŁOWIEK – NAJLEPSZA INWESTYCJA!
Centrum Medyczne Kształcenia Podyplomowego w Warszawie
PATOFIZJOLOGIA MIAŻDŻYCY I CHOROBY NIEDOKRWIENNEJ SERCA
Projekt współfinansowany przez Unię Europejską z Europejskiego Funduszu Społecznego
„Kształcenie w ramach procesu specjalizacji lekarzy deficytowych specjalności
tj. onkologów, kardiologów i lekarzy medycyny pracy”
PATOFIZJOLOGIA
MIAŻDŻYCY
I CHOROBY
NIEDOKRWIENNEJ
SERCA
Redaktor naukowy
prof. dr hab. n. med. Andrzej Beręsewicz
PATOFIZJOLOGIA MIAŻDŻYCY
I CHOROBY NIEDOKRWIENNEJ
SERCA
Redakcja naukowa
prof. dr hab. n. med. Andrzej Beręsewicz
Warszawa 2011
Przygotowanie i druk podręcznika współfinansowany przez Unię Europejską
z Europejskiego Funduszu Społecznego
AUTORZY
Prof. dr hab. med. Andrzej Beręsewicz
Dr n. med. Monika Duda
Mgr inż. Emilia Klemenska
Dr n. med. Anna Konior
Dr hab. n. med. Michał Mączewski
WYDAWCA
Centrum Medyczne Kształcenia Podyplomowego
01-813 Warszawa, ul. Marymoncka 99/103
tel. 22 56 93 700
fax 22 56 93 712
www.cmkp.edu.pl
ISBN 978-83-62110-30-8
Skład, przygotowanie do druku, druk i oprawa
Agencja Reklamowo-Wydawnicza
A. Grzegorczyk
www.grzeg.com.pl
Redaktor techniczny
Grażyna Dziubińska
Spis treści
Lista skrótów ..........................................................................................................................
I.
Miażdżyca i choroba niedokrwienna serca z lotu ptaka ....................................
7
9
Andrzej BERĘSEWICZ
I.1. Miażdżyca najczęstszym zabójcą ludzkości............................................................................
I.2. Niedokrwienie towarzyszące blaszkom miażdżycowym najczęstszym
mechanizmem „choroby sercowo-naczyniowej” ................................................................
I.3. Przekształcenia epidemiologiczne. Gdzie jest Polska? .....................................................
I.4. Czynniki ryzyka sercowo-naczyniowego .................................................................................
I.5. Ewolucyjne uwarunkowania epidemii otyłości i miażdżycy?......................................
I.6. Historia naturalna miażdżycy...........................................................................................................
II.
9
11
13
15
17
21
Energetyka serca ....................................................................................................... 27
Andrzej BERĘSEWICZ
II.1.
II.2.
II.3.
II.4.
III.
Podstawowe pojęcia i liczby.............................................................................................................
Zużycie energii, konsumpcja O2 i efektywność mechaniczna serca ......................
Podwójny produkt – nieinwazyjny wskaźnik MVO2 .........................................................
VO2max miarą obciążenia energetycznego organizmu ...............................................
27
31
31
32
Fizjologia krążenia wieńcowego ............................................................................ 35
Michał MĄCZEWSKI
III.1. Anatomia czynnościowa krążenia wieńcowego .................................................................
III.2. Determinanty fizyczne przepływu wieńcowego ................................................................
III.2.1. Zależności między oporem ciśnieniem i przepływem .......................................
III.2.2. Dystrybucja oporu wieńcowego i koncepcja fractional
flow reserve ...................................................................................................................................
III.2.3. Czynnościowy opór wieńcowy i jego regulacja .....................................................
III.2.3.1. Regulacja miogenna ..............................................................................................
III.2.3.2. Regulacja metaboliczna .......................................................................................
III.2.3.3. Regulacja śródbłonkowa .....................................................................................
III.2.3.4. Regulacja nerwowa i humoralna ...................................................................
35
36
36
38
39
40
40
41
42
3
III.2.4. Opór kompresyjny.....................................................................................................................
III.2.5. Autoregulacja przepływu wieńcowego i rezerwa wieńcowa ........................
III.3. Przepływ wieńcowy w prawej komorze ...................................................................................
III.4. Regulacja przepływu wieńcowego w wysiłku ......................................................................
III.5. Perfuzja miokardium w obecności różnych klas zwężeń
tętnic wieńcowych ................................................................................................................................
III.6. Krążenie wieńcowe w nadciśnieniu tętniczym ....................................................................
III.7 Kardiologiczny zespół X – zaburzenia mikrokrążenia wieńcowego .........................
III.8. Czynnościowe metody oceny istotności hemodynamicznej stenozy ..................
III.8.1. Scyntygrafia perfuzyjna (SPECT) .....................................................................................
III.8.2. Farmakologiczny test obciążeniowy z wazodilatatorem ..................................
III.8.3. Regionalna rezerwa wieńcowa (fractional flow reserve, FFR) ........................
III.8.4. Echokardiografia obciążeniowa........................................................................................
IV.
44
46
49
49
50
52
53
54
54
55
56
57
Przemiany substratów energetycznych w sercu ................................................. 59
Andrzej BERĘSEWICZ
IV.1. Ogólny schemat metabolizmu energetycznego ................................................................
IV.2. Transport ATP z mitochondriów do aparatu kurczliwego .............................................
IV.3. Przemiany glukozy i kwasu mlekowego...................................................................................
IV.4. Przemiany niezestryfikowanych kwasów tłuszczowych (NEFA) .................................
IV.5. Preferencje substratowe i ich wpływ na efektywność mechaniczną serca ........
IV.6. Poziomy gukozy i NEFA w surowicy regulatorami metabolizmu serca;
rola insuliny ................................................................................................................................................
IV.7. Równowaga energetyczna serca podczas wysiłku – rola kinazy AMP ..................
V.
59
61
62
63
65
66
69
Patofizjologia niedokrwienia i reperfuzji ............................................................. 73
Andrzej BERĘSEWICZ
V.1. Definicje i charakterystyka ogólna ...............................................................................................
V.2. Dwa mechanizmy niedokrwienia .................................................................................................
V.2.1. Niedokrwienie spoczynkowe ............................................................................................
V.2.2. Niedokrwienie wysiłkowe .....................................................................................................
V.3. Degradacja ATP w niedokrwieniu – regulacyjne i niekorzystne konsekwencje ...
V.4. Ból wieńcowy (angina pectoris).....................................................................................................
V.5. Zaburzenia kurczliwości mięśnia sercowego w niedokrwieniu i reperfuzji .......
V.5.1. Kurczliwość w niedokrwieniu wysiłkowym; próba dobutaminowa ...........
V.5.2. Kurczliwość w niedokrwieniu spoczynkowym; hibernowany mięsień
sercowy.............................................................................................................................................
V.5.3. Kurczliwość w reperfuzji i ogłuszenie.............................................................................
V.5.4. Lokalne odwracalne zaburzenia kurczliwości u osób z chorobą wieńcową ...
V.6. Metabolizm energetyczny serca w niedokrwieniu i reperfuzji...................................
V.6.1 Metabolizm w niedokrwieniu wysiłkowym ................................................................
V.6.2. Metabolizm w zawale ..............................................................................................................
V.6.3. Metabolizm w reperfuzji ........................................................................................................
V.7. Komórkowa kwasica i toksyczna akumulacja Ca2+ w niedokrwieniu
i reperfuzji ...................................................................................................................................................
V.7.1. Homeostaza jonów Ca2+ i H+ w niedokrwieniu i reperfuzji;
Paradoks pH ...................................................................................................................................
4
73
76
76
76
78
80
82
83
84
85
87
90
91
92
93
95
95
V.7.2. Toksyczne efekty komórkowej akumulacji Ca2+.....................................................
V.8. Mitochondria źródłem ATP, ale również ROS, apoptozy i reperfuzyjnego
uszkodzenia .............................................................................................................................................
V.8.1. Fosforylacja oksydacyjna i produkcja ATP ..................................................................
V.8.2. Mitochondria, wolne rodniki tlenowe i komórkowa ochrona
anty-rodnikowa.......................................................................................................................................
V.8.3. Mitochondria vs. apoptoza .................................................................................................
V.8.4. Mitochondrialny megakanał integratorem mechanizmów
reperfuzyjnego uszkodzenia .........................................................................................................
V.9. Śmierć komórek w niedokrwionym i reperfundowanym miokardium ...............
V.9.1. Martwica, apoptoza, zawał – definicje .........................................................................
V.9.2. Niejednorodność zaburzeń perfuzji w dorzeczu tętnicy dozawałowej ..
V.9.3. Efekty wczesnej i późnej reperfuzji ................................................................................
V.9.4. Wątpliwości dotyczące dynamiki rozwoju zawału u człowieka ...................
V.9.5. Pozawałowa przebudowa serca w erze leczenia reperfuzyjnego zawału .....
V.10. No-reflow phenomenon...................................................................................................................
V.10.1. Aspekty kliniczne no-reflow phenomenon ..........................................................
V.10.2. Reperfuzyjna dysfunkcja śródbłonkowa ..................................................................
V.10.3. Reperfuzyjny odczyn zapalny .........................................................................................
VI.
97
100
101
102
104
106
107
107
109
112
113
115
117
118
120
121
Kardioprotekcja ......................................................................................................... 129
Andrzej BERĘSEWICZ
VI.1. Krążenie pozaustrojowe i transplantacja serca ...................................................................
VI.2. Stabilna choroba wieńcowa ............................................................................................................
VI.2.1. β-blokery i iwabradyna ........................................................................................................
VI.2.2. Blokery kanału wapniowego ............................................................................................
VI.2.3. Nitrogliceryna i inne organiczne azotany .................................................................
VI.2.4. Modulatory metabolizmu ..................................................................................................
VI.3. Hartowanie serca ...................................................................................................................................
VI.3.1. Definicje.........................................................................................................................................
VI.3.2. Molekularny mechanizm hartowania ........................................................................
VI.3.3. Hartowanie w sercu człowieka ......................................................................................
VI.3.3.1. Warm up phenomenon ....................................................................................
VI.3.3.2. Dwie następujące po sobie próby wysiłkowe.....................................
VI.3.3.3. Angina przed zawałem ......................................................................................
VI.3.3.4. Hartowanie u pacjentów z zawałem.........................................................
VI.3.3.5. Hartowanie w okołoproceduralnym uszkodzeniu miokardium ....
VII.
131
131
132
134
135
137
139
139
141
142
142
142
142
143
144
Patofizjologia miażdżycy i ostrych zespołów wieńcowych ............................... 149
Andrzej BERĘSEWICZ, Emilia KLEMENSKA
VII.1. Wrodzona odporność i jej udział w mechanizmie miażdżycy ..................................
VII.1.1. Repetytorium z mechanizmów wrodzonej odporności ...............................
VII.1.2. Receptory zmiatające biorące udział w powstawaniu miażdżycy ..........
VII.1.3. Receptory TLR ...........................................................................................................................
VII.2. Mechanizm komórkowy ogniskowych naczyniowych zmian miażdżycowych....
VII.3. Zmiany naczyniowe towarzyszące chorobie wieńcowej .............................................
150
150
154
157
158
160
5
VII.3.1. Klasy stenoz i ich wpływ na symptomatologię stabilnej dusznicy ..........
VII.3.2. Blaszki koncentryczne i ekscentryczne .....................................................................
VII.3.3. Ogniskowe vs. uogólnione rozprzestrzenienie zmian.....................................
VII.3.4. Przebudowa ściany naczyniowej ................................................................................
VII.3.5. Dysfunkcja śródbłonkowa.................................................................................................
VII.4. Ostre zespoły wieńcowe (OZW) ...................................................................................................
VII.4.1. Badania autopsyjne u osób zmarłych w wyniku OZW ....................................
VII.4.2. Czteroczynnikowy model mechanizmu OZW .....................................................
VII.4.2.1. Niestabilna blaszka miażdżycowa ............................................................
VII.4.2.2. Niestabilny układ krzepnięcia .....................................................................
VII.4.2.3. Bodziec uszkadzający ......................................................................................
VII.4.2.4. Systemowe zapalenie i „niestabilny” pacjent......................................
VIII.
161
161
161
162
163
164
164
165
167
171
172
173
Naczyniowy stres oksydacyjny ............................................................................... 179
Emilia KLEMENSKA
VIII.1. Słowniczek pojęć i terminów wolnorodnikowych ..........................................................
VIII.2. ROS w naczyniach ..............................................................................................................................
VIII.3. Rola wolnorodnikowej modyfikacji LDL w patomechanizmie miażdżycy .......
VIII.4. Próby kliniczne z interwencjami anty-rodnikowymi ......................................................
IX.
179
181
184
184
Śródbłonek naczyniowy i jego dysfunkcja .......................................................... 187
Michał MĄCZEWSKI, Anna KONIOR
IX.1. Budowa i role biologiczne śródbłonka......................................................................................
IX.2. Tlenek azotu – główny mediator śródbłonkowy ................................................................
IX.3. Regeneracja śródbłonka ...................................................................................................................
IX.4. Naczynio-rozkurczające i przeciwmiażdżycowe działanie śródbłonka.................
IX.4.1. Śródbłonkowa regulacja przepływu tkankowego ...............................................
IX.4.2. Działanie przeciwzapalne i przeciwmiażdżycowe śródbłonka ....................
IX.4.3. Działanie przeciwkrzepliwe i prokoagulacyjne śródbłonka...........................
IX.5. Kliniczne metody pomiaru czynności śródbłonka ...........................................................
IX.6. Choroby i stany kliniczne z dysfunkcją śródbłonkową ....................................................
IX.7. Mechanizm klinicznych postaci dysfunkcji śródbłonkowej .........................................
IX.7.1. Nadmierna inaktywacja NO ...............................................................................................
IX.7.2. Zmniejszone wytwarzanie NO .........................................................................................
IX.8. Interwencje lecznicze o działaniu śródbłonkowym ..........................................................
187
189
193
193
193
194
195
195
197
199
199
200
201
X.
Zespół kardiometaboliczny ..................................................................................... 205
6
Monika DUDA
X.1. Definicja i kryteria diagnostyczne..................................................................................................
X.2. Epidemiologia ............................................................................................................................................
X.3. Rola tkanki tłuszczowej w rozwoju zespołu kardiometabolicznego ......................
X.3.1. Krótka charakterystyka tkanki tłuszczowej ................................................................
X.3.2. Otyłość trzewna i ektopowe gromadzenie lipidów .............................................
X.4. Mechanizm insulinooporności i zaburzeń gospodarki węglowodanowej .........
X.5. Mechanizm nadciśnienia tętniczego...........................................................................................
X.6. Związek miażdżycy z zespołem kardiometabolicznym ...................................................
X.7. Zaburzenia czynności skurczowej serca w zespole kardiometabolicznym.........
205
207
208
208
210
211
211
212
213
Lista skrótów
ADMA
ADP
AGE
AMP
AMPK
ATP
BH4
CABG
CNS
CoA
CPT-I
CrP
CRP
DAMPs
ΔΨm
ecSOD
ET-1
GLUT
GPZ
HN
HR
H2O2
ICAM-1
LDL
LOX-1
MCP-1
M-CSF
– niesymetryczna dimetyloarginina (endogenny inhibitor NOS)
– adenozynodifosforan
– zaawansowane produkty glikacji białek (Advanced Glication Products)
– adenozynomonofosforan
– kinaza zależna od AMP
– adenozynotrifosforan
– tetrahydrobiopteryna (kofaktor NOS)
– pomostowanie tętnic wieńcowych (coronary artery bypass grafting)
– choroba niedokrwienna serca
– koenzym A CPK – kinaza kreatynowa
– transferaza karnityno-palmitynowa I (Carnitine-Palmitate Transferase I)
– fosforan kreatyny (Creatine Phosphate)
– białko C-reaktywne (C-reactive protein)
– endogenne alarminy (damage-associated molecular patterns)
– mitochondrialny gradient elektrochemiczny protonów
– zewnątrzkomórkowa izoforma dyzmutazy ponadtlenkowej
– endotelina 1
– błonowy transporter glukozy (GLUcose Transporter)
– gałąź przednia zstępująca lewej tętnicy wieńcowej
– hartowanie niedokrwieniem (ischemic preconditioning)
– hartowanie reperfuzją (ischemic postconditioning)
– nadtlenek wodoru
– międzykomórkowa cząstka adhezyjna-1 (Inter-Cellular Adhesion Molecule-1)
– lipoproteiny o małej gęstości
– receptor oksydowanych LDL (lectine-like oxidized LDL receptor)
– białko chemotaktyczne dla monocytów (monocyte chemoattractant/chemotactic protein-1
– czynnik stymulujący wzrost kolonii makrofagów (macrophage colony stimulating factor)
7
MDA
mitoK-ATP
mtALDH
mPTP
MVO2
NEFA
NF-кB
NO
NOS
– dialdehyd malonylowy (marker peroksydacji lipidów)
– mitochondrialny kanał potasowy zależny od ATP
– mitochondrialna dehydrogenaza alkoholowa
– megakanał mitochondrialny (mitochondrial permeability transition pore)
– konsumpcja tlenu przez miokardium
– niezestryfikowane (wolne) kwasy tłuszczowe (Nonesterified Fatty Acids)
– czynnik jądrowy (transkrypcyjny) кB (nuclear factor кB)
– tlenek azotu
– syntaza tlenku azotu – występują trzy izoformy: śródbłonkowa (eNOS), neuronalna (nNOS), i indukowana (iNOS)
– tlen cząsteczkowy
O2
O2–
– anionorodnik ponadtlenkowy
OH–
– rodnik wodorotlenowy
ox-LDL
– oksydacyjnie zmodyfikowane LDL
OZW
– ostre zespoły wieńcowe
PAI-1
– inhibitor tkankowego aktywatora plazminogenu (tPA)
PAMPs
– egzogenne alarminy (pathogen-associated molecular patterns)
PCI
– przezskórna plastyka wieńcowa (percutaneous coronary intervention)
PDGF
– płytko-pochodny czynnik wzrostu (platelet derived growth factor)
PDH
– dehydrogenaza pirogronianowa (Pyruvate Dehydrogenase)
Pi
– fosfor nieorganiczny
PRR
– receptory alarmin czyli PAMPs i DAMPs (Pattern Recognition Receptors)
ROS
– reaktywne formy tlenu (Reactive Oxygen Species), np. O2–, H2O2 i OH–
SOD
– dyzmutaza ponadtlenkowa (Superoxide Dismutase)
SR-A i SR-B – receptory wymiatające A i B (scavenger receptors)
TLR
– receptory TLR (Toll-like receptors) (subklasa receptorów PRR)
TNF-α
– czynnik martwicy nowotworów (tumor necrosis factor-α)
tPA
– tkankowy aktywator plazminogenu
VCAM-1 – cząstka adhezyjna-1 komórek naczyniowych (vascular cell adhesion molecule-1)
ZKM
– zespół kardio-metaboliczny
8
I. Miażdżyca i choroba niedokrwienna serca
z lotu ptaka
Andrzej BERĘSEWICZ
I.1. Miażdżyca najczęstszym zabójcą ludzkości
W badaniach epidemiologicznych, termin choroba sercowo-naczyniowa (Ch.S.N.) (cardiovascular disease, CVD) obejmuje wszystkie choroby serca i naczyń, w tym choroby tętnic
i żył. Poza Afryką Subsaharyjską, choroby serca i naczyń są obecnie najczęstszą przyczyną
zgonów na świecie. Zgodnie ze statystykami WHO, w 2005 roku liczba zgonów na świecie
wynosiła 58 mln, w tym było 17,5 mln zgonów z powodu „choroby sercowo-naczyniowej”, co
stanowiło 30,2% wszystkich zgonów (umieralność sercowo-naczyniowa 30,2%). Drugą, co do
częstości (9,3%), przyczyną zgonów na świecie były choroby zakaźne (HIV – 2,8 mln, gruźlica – 1,8 mln, malaria 0,8 mln). Udział chorób serca i naczyń w ogólnej statystyce zgonów
różni się jednak drastycznie pomiędzy różnymi rejonami świata, w zależności od stopnia ich
rozwoju ekonomiczno-cywilizacyjnego. Dla przykładu, zgony te stanowiły 10%, 58%, 38,5%
i 35,3% wszystkich zgonów, odpowiednio, w Afryce Subsaharyjskiej, Europie Wschodniej +
Rosji, krajach wysoko uprzemysłowionych ogólnie oraz w USA (ryc. 1.1.).
Niezależnie od regionu świata, o śmiertelności z powodu Ch.S.N decydują przede wszystkim zaburzenia, których podłożem jest miażdżyca tętnic (ryc. 1.2.). Prawdopodobnie z tego
powodu, wielu autorów używa terminu choroba sercowo-naczyniowa jako synonimu tychże
miażdżyco-pochodnych chorób. Istotę problemu bardziej precyzyjnie oddają jednak anglojęzyczne terminy „atherosclerotic cardiovascular disease” (vide Braunwald’s Heart Disease,
IX edition) lub „atherothrombotic vascular disease” [2]. Dość zbliżony zakres znaczeniowy
ma funkcjonujący w Polsce termin „aterotromboza” ale, w istocie, brak jest dobrego polskiego odpowiednika (miażdżycopochodna choroba sercowo-naczyniowa, M.Ch.S.N.?) W tych
terminologicznych zabiegach chodzi o myśl, że miażdżyco-pochodne zespoły chorobowe są
bardzo częste, są wywoływane przez podobne czynniki, mają wspólny komórkowy mechanizm i prawdopodobnie mogą być podobnie leczone.
9
Ryc. 1.1. Umieralność z powodu chorób serca i naczyń (cardiovascular disease) w różnych rejonach
i krajach świata uszeregowanych wg ich rosnącego statusu ekonomiczno-cywilizacyjnego. Wraz z rozwojem ekonomicznym rośnie udział tych chorób w ogólnej statystyce zgonów, ale od pewnego poziomu
rozwoju, umieralność sercowo-naczyniowa ponownie maleje. Dane z ref. [1].
Ryc. 1.2. Statystyka zgonów z powodu różnych postaci choroby sercowo-naczyniowej w krach cywilizacji zachodniej oraz w Europie Wschodniej i dawnym ZSRR. W obu rejonach główną przyczyną zgonów
„sercowo-naczyniowych” są stany chorobowe będące kliniczną manifestacją miażdżycy, w tym zwłaszcza choroba niedokrwienna serca (CNS) i udar mózgu. W klasie „Inne” mieszczą się zgony z powodu
niewydolności serca i arytmii, ale CNS jest przyczyną ~70% przypadków niewydolności serca i 80%
nagłych śmierci sercowych. (wg Braunwald’s Heart Disease, IX edition).
10
I.2. Niedokrwienie towarzyszące blaszkom miażdżycowym najczęstszym
mechanizmem „choroby sercowo-naczyniowej”
Miażdżyca (atherosclerosis, arteriosclerosis); jest przewlekłym procesem zapalnym
dotyczącym aorty i tętnic średniej wielkości (wieńcowe nasierdziowe, szyjne, nerkowe biodrowe, kreskowe). Z histologicznego punktu widzenia, proces polega na gromadzeniu się
w przestrzeni pomiędzy śródbłonkiem i warstwą mięśniową tętnicy (tunica intima) początkowo – złogów składających się z makrofagów, lipoprotein o małej gęstości (LDL), komórek
piankowatych (czyli makrofagów obładowanych oksydowanymi LDL) i pozakomórkowych
skupisk cholesterolu. Powstające w ten sposób, i widoczne gołym okiem na wewnętrznej powierzchni naczynia, pasma tłuszczowe (ang. fatty streaks) są najwcześniejszą postacią zmian
miażdżycowych i znakiem rozpoznawczym miażdżycy w badaniach autopsyjnych. Z czasem
zawartość pasm tłuszczowych zostaje uzupełniona o elementy włókniste tkanki łącznej, które
przerastając i otaczając pierwotne ognisko zapalne, separują je od reszty naczynia. Powstająca w ten sposób zaawansowana zmiana miażdżycowa określana jest terminem „blaszka
miażdżycowa” (atheromatous plaque) (ryc. 1.3.). Z wiekiem rośnie zarówno liczba blaszek
jak i stopień ich wewnętrznego zróżnicowania. U tej samej osoby mogą występować zmiany
miażdżycowe w różnych fazach rozwoju.
Ryc. 1.3. Struktura histologiczna blaszki miażdżycowej. Prawidłowa tętnica składa się, od wnętrza naczynia: z jednokomórkowej warstwy komórek śródbłonka, błony mięśniowej (tunica media) i przydanki
(adventitia). Proces miażdżycowy polega na tym, że między śródbłonkiem a błoną mięśniową dużych
tętnic (tunica intima) dochodzi do ogniskowego gromadzenia się: (i) komórek zapalnych (głównie makrofagów powstałych z przekształcenia monocytów); (ii) lipoprotein o małej gęstości (LDL) oraz (iii)
przekształconych komórek mięśni gładkich produkujących elementy tkanki łącznej. Makrofagi fagocytujące LDL (komórki piankowate) obumierają, a ich lipidowa zawartość gromadzi się pozakomórkowo,
tworząc jądro lipidowe blaszki. Tkanka łączna otaczająca jądro lipidowe stanowi główny składnik pokrywy blaszki miażdżycowej.
Główne kliniczne manifestacje miażdżycy związane są dopiero z obecnością blaszek
w tętnicach, i związanymi z blaszkami zaburzeniami perfuzji tkankowej i niedokrwieniem.
Stąd manifestacje chorobowe miażdżycy tętnic wieńcowych określane są terminem choroba
niedokrwienna serca (CNS) (ischemic heart disease, choroba wieńcowa). O symptomatologii
klinicznej związanej z blaszkami decydują dwie ich właściwości:
11
1. Niektóre blaszki powoli wpuklają się do światła naczynia i zwężając je ograniczają rezerwę
wazodilatacyjną w obszarze za zwężeniem. W sercu, przewlekłe zwężenie (stenoza) światła
jednej lub wielu nasierdziowych tętnic wieńcowych przez blaszki skutkuje zmniejszeniem
rezerwy wieńcowej w dorzeczu zwężonych tętnic, co jest podstawą patomechanizmu wysiłkowego niedokrwienia i zespołu znanego jako stabilna choroba wieńcowa (aspekt „luminologiczny” miażdżycy i blaszki miażdżycowej);
2. Śródbłonek i pokrywa łącznotkankowa blaszki mogą ulegać erozji/rozerwaniu. Umożliwia to kontakt krwi z trombogenną zawartością blaszki i może prowadzić do powstania
wewnątrznaczyniowego zakrzepu. Od tego etapu, objawy kliniczne, jeżeli w ogóle obecne,
związane są raczej z zakrzepem a nie blaszką, per se. Powstawanie zakrzepu w miażdżycowo zmienionej tętnicy określane jest w literaturze anglojęzycznej terminem atherothrombosis. Zakrzep powstały w tym mechanizmie może skutkować częściowym lub całkowitym zamknięciem światła tętnicy i nagłym ograniczeniem/zahamowaniem spoczynkowego przepływu krwi w jej dorzeczu. W przypadku serca, konsekwencją zaburzeń perfuzji
wtórnych do „aterotrombozy” mogą być cztery zespoły chorobowe (nagła śmierć sercowa,
niestabilna choroba wieńcowa, zawał serca bez uniesienia załamka ST i zawał serca z uniesieniem załamka ST) znane jako ostre zespoły wieńcowe (OZW).
O miażdżycy dodatkowo wiadomo, że:
1. występuje praktycznie w całej, lub prawie całej, dorosłej populacji mieszkańców krajów
cywilizacji zachodniej (rozdz. I.6.);
2. zaczyna się często już w życiu płodowym lub wczesnym dzieciństwie (rozdz. I.6.);
3. staje się źródłem objawów klinicznych, które ujawniają się – u mężczyzn najczęściej dopiero w piątej dekadzie życia lub później, a u kobiet – w okresie po menopauzie (ryc. 1.4.);
Ryc. 1.4. Historia naturalna miażdżycy. Zmiany miażdżycowe w tętnicach pojawiają się już w życiu
płodowym lub wczesnym dzieciństwie. Natomiast manifestacje kliniczne choroby, najczęściej związane
z obecnością blaszek miażdżycowych, ujawniają się dopiero w drugiej połowie życia.
4. aorta jest miejscem gdzie zmiany występują najwcześniej i są najrozleglejsze (rozdz. I.6.),
jednakże miażdżyca jest chorobą całego układu tętniczego. Świadczy o tym fakt, że stwierdzenie zmian miażdżycowych w jednym obszarze naczyniowym zwiększa prawdopodo12
bieństwo zaburzeń pochodzących z innych obszarów [3,4]. Dla przykładu, osoby z obniżonym wskaźnikiem „kostka-ramię” (nieinwazyjny wskaźnik zaawansowania zmian miażdżycowych w tętnicach obwodowych) maję zwiększone ryzyko zawału serca, incydentów
niedokrwienia mózgu (TIA) i udaru mózgu [5]. Podobnie u osób z udarem mózgu rośnie
prawdopodobieństwo wystąpienia CNS [5].
5. choć miażdżyca jest procesem całego układu tętniczego, z niejasnych powodów, najczęstszą
manifestacją miażdżycy są CNS i udar mózgu (w USA występowanie odpowiednio u 5,6%
i 2,2% populacji). Także umieralność z powodu CNS jest w przybliżeniu dwa razy częstsza
od umieralności z powodu udaru (ryc. 1.2.). Kolejne, co do częstości występowania, są
zaburzenia związane z miażdżycą aorty (tętniaki, rozwarstwienia), tętnicy nerkowej, oraz
tętnic kończyn dolnych [2].
6. o miażdżycy tętnic wieńcowych i spowodowanej nią CNS wiadomo, że jest przyczyną ~70%
przypadków niewydolności serca i ~80% nagłych śmierci sercowych.
I.3. Przekształcenia epidemiologiczne. Gdzie jest Polska?
Analiza historycznych szacunków i współczesnych bardziej wiarygodnych statystyk sugeruje,
że rozwój ekonomiczno-cywilizacyjny ludzkości można podzielić na cztery etapy ważne z punktu
widzenia udziału zgonów sercowo-naczyniowych w ogólnej statystyce zgonów (ryc. 1.5.). Periodyzacja ta znana jest w anglojęzycznej literaturze jako „epidemiological transitions” [1].
Ryc. 1.5. Cztery etapy przekształceń epidemiologicznych, w wyniku, których udział zgonów sercowonaczyniowych w ogólnej statystyce zgonów rósł przez wieki, a ostatnio w krajach uprzemysłowionych
spada. Najnowsze statystyki sugerują, że ten trend spadkowy ulega zahamowaniu, a nawet, że umieralność sercowo-naczyniowa znowu zaczyna rosnąć (era otyłości i nieaktywności?).
13
Od zarania dziejów aż do przełomu osiemnastego i dziewiętnastego wieku, główną przyczyną zgonów były choroby zakaźne i stany będące skutkiem niedożywienia (I Etap – era
zaraz i głodu). Długość życia wynosiła wtedy ~35 lat, a zgony sercowo-naczyniowe stanowiły <10%. Przyjmuje się, że Afryka Subsaharyjska, gdzie główną przyczyną zgonów są ciągle
choroby zakaźne, a zgony sercowo naczyniowe stanowią ~10% ciągle znajduje się na I etapie
przekształceń epidemiologicznych (ryc. 1.1.)
II Etap – era nawracających pandemii; w dziewiętnastym wieku i na początku dwudziestego, wraz z rewolucją przemysłową i urbanizacją, poprawił się stan odżywienia i higieniczny
ludności (przynajmniej w USA i Europie Zachodniej), co skutkowało wydłużeniem przewidywanej długości życia do ~50 lat. W tym czasie zmalała liczba zgonów z powodu niedożywienia, a udział zgonów sercowo-naczyniowych wzrósł do 10–35%. Przyjmuje się, że kraje
rozwijające się (np. Chiny i Indie), są obecnie na II etapie przekształceń epidemiologicznych
(ryc. 1.1.).
III. Etap – era chorób degeneracyjnych i chorób spowodowanych działalnością człowieka; w USA etap ten obejmował lata 30–60 ubiegłego wieku, a w Europie Zachodniej miał
on miejsce około 10 lat później. Jest to era postępującej urbanizacji, wzrostu stopy życiowej,
skutecznej walki z infekcjami i wydłużenia życia do >60 lat. Ale równocześnie jest to era lawinowego narastania różnych szkodliwości (~60% Amerykanów paliło wtedy papierosy, wysoko-tłuszczowa i wysoko-kaloryczna dieta, brak ruchu) skutkujących rozwojem miażdżycy. Na
tym etapie, udział zgonów sercowo-naczyniowych rośnie do >60% a najczęstszą ich przyczyną staje się CNS. Jak sugeruje ryc. 1.1. na tym etapie przekształceń znajduje się obecnie Rosja
i republiki post-radzieckie.
IV. Etap – era dobrobytu i sukcesów prewencji miażdżycy (<30% Amerykanów pali
obecnie papierosy), zwiększonej efektywności leczenia (np. zawału serca), ale równocześnie
zwiększonego występowania „opóźnionych” chorób degeneracyjnych (np. niewydolności
serca). Na tym etapie znajdują się USA i inne wysoko uprzemysłowione kraje świata. W krajach tych spodziewana długość życia wynosi >70 lat, a udział zgonów sercowo-naczyniowych
od kilku lat systematycznie maleje o ~3% rocznie i aktualnie wynosi <40% [1] (ryc. 1.1.).
Przyjmuje się, że o tym korzystnym trendzie zadecydowała głównie redukcja czynników ryzyka i w mniejszym stopniu nowe metody leczenia [6].
Hipotetyczny V etap – era otyłości i nieaktywności fizycznej; w USA i Europie Zachodniej spotykają się obecnie dwa niepokojące zjawiska epidemiologiczne. Z jednej strony, od
jakiegoś czasu nie obserwuje się już postępującego spadku liczby palaczy w populacji i dodatkowo ustabilizował się poziom wykrywalności i częstości leczenia nowych przypadków
nadciśnienia tętniczego (redukcja ważnych czynników ryzyka osiągnęła kres możliwości).
Z drugiej strony, rośnie epidemia otyłości, cukrzycy typu II i zespołu kardio-metabolicznego
(rozdz. X). Powstaje wobec tego pytanie, czy nowoczesne terapie są w stanie skompensować
te niekorzystne zjawiska epidemiologiczne i czy możliwe będzie kontynuowanie dotychczasowego trendu spadkowego umieralności sercowo-naczyniowej. W tym kontekście, ogromny
niepokój wzbudziły niedawne doniesienia o „spłaszczeniu” i lekkim wzroście trendu umieralności sercowo-naczyniowej u mężczyzn i kobiet w wieku 35–44 lat w USA [7]. Podobnie, konkluzja z systematycznych badań autopsyjnych, obejmujących okres od 1981 roku do
2004 roku i dotyczących nagle zmarłych rezydentów miasta Olmsted w stanie Minnesota,
14
USA, jest taka, że era systematycznego spadku rozległości i zaawansowania miażdżycowych
zmian w tętnicach wieńcowych skończyła się około 1995 roku, i że od 2000 roku nasilenie
tych zmian ponownie rośnie [8].
Ryc. 1.6. Trendy umieralności sercowo-naczyniowej w różnych krajach Europy. Posługując się periodyzacją przedstawioną na ryc. 1.5., można zaryzykować twierdzenie, że: (i) Rosja, Ukraina i Bułgaria są na
III etapie przekształceń epidemiologicznych; (ii) Polska od lat dziewięćdziesiątych ubiegłego wieku jest
na etapie IV; (iii) kraje Europy Zachodniej osiągnęły etap IV znacznie wcześniej niż Polska i dlatego ich
statystyki umieralności sercowo-naczyniowej są lepsze niż w Polsce oraz (iv) Francja, ze swymi bardzo
niskimi wskaźnikami umieralności jest fenomenem wśród krajów uprzemysłowionych, co znane jest
w literaturze jako francuski paradoks (french paradox).
W Polsce dopiero niedawno wkroczyliśmy w okres systematycznego spadku trendu
umieralności sercowo-naczyniowej (ryc. 1.6.), co stwarza nadzieję na kontynuację tego trendu jeszcze w najbliższej przyszłości.
I.4. Czynniki ryzyka sercowo-naczyniowego
Jedną z dróg do poznania przyczyn miażdżycy są badania epidemiologiczne. Pierwsze poważne badanie tego typu rozpoczęło się w 1948 roku i dotyczyło mieszkańców
Farmingham, miasta satelitarnego Bostonu, USA (Farmingham Heart Study). Do badania
włączono 5209 stałych mieszkańców Farmingham w wieku 30–62 lat i od tej pory aż do
dzisiaj osoby te są poddawane badaniom w odstępach dwuletnich. W latach siedemdziesiątych ubiegłego wieku obserwacją objęto dodatkowo 5153 dorosłych potomków pierwszych uczestników badania. W oparciu o te wieloletnie obserwacje badacze ustalili listę
czynników, które predysponują do rozwoju klinicznych powikłań miażdżycy (Tab. 1.1.)
i które następnie opatrzyli terminem cardiovascular risk factors (czynniki ryzyka sercowo-naczyniowego).
Czynniki ryzyka są to stany, które zwiększają statystyczne prawdopodobieństwo wystąpienia jakiejś formy choroby sercowo-naczyniowej przynajmniej o 100% (wzrost prawdopodobieństwa <100% jest uznawany za słaby związek). Należy jednak pamiętać, że z epidemiologicznych badań wynikają wnioski jedynie co do korelacji statystycznych, a nie związków
15
przyczynowo-skutkowych między zjawiskami. Nie dziwi wobec tego fakt, że liczne osoby bez
czynników ryzyka, mimo wszystko rozwijają chorobę sercowo-naczyniową.
Tabela 1.1. Czynniki ryzyka i predykatory rzadszego występowania choroby sercowo-naczyniowej
Modyfikowalne czynniki ryzyka
– podwyższone ciśnienie krwi
– hipercholerterolemia
– lipoproteina (a)
– palenie papierosów
– otyłość brzuszna
– nietolerancja glukozy
– cukrzyca
– fibrynogen
– przerost lewej komory
– zażywanie kokainy
– typ osobowości A
Niemodyfikowalne czynniki ryzyka
– wiek
– płeć
– uwarunkowania genetyczne
„Predykatory” rzadszego występowania
– cholesterol HDL
– wysiłek fizyczny
– estrogeny
– alkohol w umiarkowanych ilościach
Kryteria uznania jakiegoś stanu za czynnik ryzyka są następujące:
1. Silny związek statystyczny czynnika z chorobą (wzrost prawdopodobieństwa wystąpienia
choroby o >100%);
2. Związek powinien być konsekwentny (występowanie niezależnie od wieku, płci, rasy
i ośrodka prowadzącego badanie);
3. Związek powinien mieć sens biologiczny;
4. Związek powinien być „reprodukowalny” w warunkach eksperymentalnych;
5. Leczenie skutkujące redukcją czynnika ryzyka powinno skutkować spadkiem prawdopodobieństwa wystąpienia choroby;
6. Należy wykazać, że czynnik da się sklasyfikować jako tzw. niezależny czynnik ryzyka wg
kryteriów statystycznych (niezwiązany z występowaniem innych czynników).
Jak wynika z dalszych rozdziałów, prawdopodobnie te same czynniki ryzyka biorą
udział w inicjowaniu wczesnych form procesu miażdżycowego (rozdz. I.6.) jak i w procesie destabilizacji już uformowanych blaszek miażdżycowych (rozdz. VII). Prawdopodobnie
dlatego ryzyko występowania choroby sercowo-naczyniowej można zmniejszyć poprzez redukcję czynników ryzyka nawet, jeżeli postępowanie prewencyjne wdrażane jest stosunkowo
późno, nawet już po wystąpieniu klinicznych objawów choroby [9]. Dane na temat wczesnego
i powszechnego występowania miażdżycy sugerują potrzebę możliwie wczesnej prewencji.
Wiadomo, że skuteczność prewencji pierwotnej w chorobie wieńcowej jest tym większa im
wcześniej zostaje wdrożona. Nie dziwi, wobec tego opinia, że prewencja choroby sercowo-naczyniowej jest, w istocie, problemem pediatrycznym, a nie tylko kardiologicznym [10].
Trwają poszukiwania jakiegoś wspólnego elementu patogenetycznego czynników ryzyka.
Jego identyfikacja przybliżyłaby ostateczne poznanie mechanizmu aterogenezy. Wydaje się,
że, przynajmniej w przypadku takich popularnych czynników ryzyka jak hypercholesterolemia, cukrzyca, nadciśnienie tętnicze i otyłość, takimi wspólnymi elementami są zwiększona
naczyniowa produkcja reaktywnych form tlenu (rozdz. VIII) oraz zaburzenia czynności i fenotypu śródbłonka naczyniowego (rozdz. IX).
16
I.5. Ewolucyjne uwarunkowania epidemii otyłości i miażdżycy?
Jedna z hipotez tłumaczących obecną epidemię otyłości (głównego obecnie czynnika ryzyka sercowo-naczyniowego) zakłada, że przyczyną problemu jest konflikt między genotypem Homo sapiens ukształtowanym 10–40 tys. lat temu w trudnej życiowo epoce kamiennej
i dobrobytem obecnej epoki kosmicznej [11-14]. Możliwe są dwie osie konfliktu:
1. genotyp człowieka epoki kamiennej ukształtował się prawdopodobnie w reakcji na powtarzające się okresy głodu – obecnie wobec nieograniczonej podaży pokarmu przyczynia się
on do niepotrzebnego gromadzenia substratów energetycznych i epidemii otyłości;
2. do niedawna, zdobywanie pokarmu było nieodłącznie związane z wysiłkiem fizycznym,
który jest prawdopodobnie genetycznie uwarunkowanym regulatorem procesów metabolicznych – obecnie tego regulatora jesteśmy pozbawieni, gdyż zdobywanie pokarmu nie
wymaga wysiłku fizycznego i generalnie mało się ruszamy.
Życie myśliwego-zbieracza epoki kamiennej składało się z następujących po sobie okresów sytości i głodu (ryc. 1.7.). Jego aktywność fizyczna sprowadzała się do zdobywania pokarmu. Sukces był okazją do ucztowania, względnej bezczynności, ale także do intensywnego
gromadzenia zapasów energetycznych na „czarną godzinę”. Ponieważ nie umiano przechowywać żywności, dopiero głód mobilizował człowieka do ponownego poszukiwania pokarmu. Bywało to trudne i okresy głodu (i chłodu) często się wydłużały. W tej sytuacji, większą
szansę przeżycia i wydania potomstwa mieli osobnicy wyposażeni w geny (cechy) umożliwiające przetrwanie długich okresów głodu. W tym kontekście, dyskutowane są dwa ważne
przystosowania pierwotnych myśliwych-zbieraczy:
1. Posiedli oni zdolność efektywnego gromadzenia glikogenu i triglicerydów w okresie sytości i bardzo oszczędnego gospodarowania nimi w okresie głodu. Postuluje się, że za taki
korzystny profil metaboliczny odpowiedzialny jest hipotetyczny zespół genów „gospodarnych” („oszczędzających”, ang. thrifty genes) [11,12] i że „gospodarny” genom przetrwał
do obecnych czasów i jest jedną z przyczyn dzisiejszej epidemii otyłości. Jednakże próby
identyfikacji „gospodarnych” genów się dotąd nie powiodły.
(A)
(B)
Ryc. 1.7. Cykl sytości-głodu i fizycznej aktywności, który prawdopodobnie ukształtował genotyp człowieka epoki kamiennej (A) oraz szkodliwe zdrowotnie przerwanie tych naturalnych cykli w obecnym
świecie (B). „Gospodarne magazynowanie” jest ciągle obecne, lub nawet nasilone ze względu na nieograniczoną podaż pokarmu. Nie występują okresy głodu i brak jest intensywnych wysiłków opróżniających
mięśniowe magazyny glikogenu (G) i triglicerydów (TG). To z kolei sprawia, że nie jest aktywowana
kinaza białkowa aktywowana przez AMP (AMPK) i nie jest aktywowany błonowy transporter glukozy
(GLUT4) w mięśniach szkieletowych.
17
2. Wykształcili oni mechanizmy zapewniające zaopatrzenie mózgu w glukozę w czasie głodu.
Wiadomo, że: (i) glukoza jest głównym substratem energetycznym mózgu; (ii) mózg nie
ma zdolności gromadzenia glukozy i jej podaż do mózgu musi być nieprzerwana oraz (iii)
koszt energetyczny pracy mózgu stanowi aż 20–25% całkowitego spoczynkowego zużycia
energii człowieka (u niższych ssaków ~5%) więc podaż glukozy do mózgu musi być bardzo duża [15]. W okresie po-posiłkowym glukoza we krwi pochodzi głównie z jej wchłaniania jelitowego. Następnie, w 70–80% jest ona wychwytywana przez mięśnie szkieletowe, co pokazuje jak aktywnym konsumentem glukozy są mięśnie. Wraz z upływem
czasu od posiłku, poziomy glukozy i insuliny we krwi maleją, co ma trzy ważne konsekwencje metaboliczne; (i) Rośnie uwalnianie niezestryfikowanych kwasów tłuszczowych
(NEFA, wolnych kwasów tłuszczowych) z tkanki tłuszczowej (rozdz. IV.6) i stają się one
podstawowym substratem energetycznym dla wszystkich narządów z wyjątkiem mózgu;
(ii) Następuje aktywacja procesu syntezy glukozy (glukoneogeneza) i rozkładu glikogenu (glikogenoliza) w wątrobie i wątroba staje się głównym źródłem glukozy obecnej we
krwi i dostępnej dla mózgu; (iii) Narządy konkurują z sobą o glukozę. W tej konkurencji
mózg musiałby przegrać z mięśniami szkieletowymi, gdyby ich zdolność wychwytywania glukozy utrzymywała się na normalnym poziomie. Tak się jednak nie dzieje, gdyż
wraz z przedłużaniem się okresu głodu, w mięśniach szkieletowych (i innych tkankach)
spada ekspresja błonowych transporterów glukozy i tkanki stopniowo tracą zdolność jej
wychwytywania (w badaniach czynnościowych widoczne to jest jako insulinooporność).
Sprawia to, że wątrobowa glukoza przechodzi do mózgu, a nie do mięśni, co umożliwia
sprawne funkcjonowanie mózgu (historycznie zwiększało to szansę na zdobycie pokarmu). Rzeczywiście, nawet w trakcie przedłużającego się głodu z towarzyszącym wysiłkiem, glikemia jest względnie stała.
Z powyższych rozważań wynika, że dla myśliwych-zbieraczy było korzystne by zdolność
wychwytywania glukozy przez ich mięśnie była możliwie duża w okresie sytości (bo sprzyjało
to magazynowaniu) i możliwie mała w okresie głodu (bo umożliwiało to funkcjonowanie
mózgu). Ewolucjoniści spekulują, że mechanizm, który sprawiał, że po okresie głodu, w czasie ucztowania dochodziło do normalizacji metabolizm glukozy, musiał być ściśle powiązany
z sukcesem pokarmowym, gdyż inaczej wiązałoby się to z utratą sprawności mózgu. Różne
argumenty sugerują, że bodźcem przywracającym normalny transport glukozy po okresie
głodu był wysiłek fizyczny, ale o natężeniu tak dużym by spowodować opróżnienie mięśniowych magazynów glikogenu (i być może triglicerydów) i prawdopodobnie spowodować aktywację pewnych genów. Sens ewolucyjny tego typu mechanizmu polegałby na tym, że duże
nagłe wysiłki opróżniające mięśniowe magazyny substratów energetycznych dawały większą
gwarancję zdobycia pokarmu niż wysiłki małe. Geny, o których tu mowa, zostały umownie nazwane „genami aktywności fizycznej (activity genes) [13,14]. Ich specyfika miała by
polegać na tym, że poziom ich ekspresji byłby regulowany poprzez aktywność ruchową i że
byłyby one regulatorem ważnych procesów życiowych. Hipoteza zakłada, że dziedziczymy
po naszych przodkach genetycznie uwarunkowaną potrzebę aktywności ruchowej i że jedną
z konsekwencji jej braku może być współczesna epidemia zaburzeń metabolizmu glukozy
(insulinooporność). Tłumaczyło by to dlaczego pro-zdrowotne działanie treningu fizycznego
ujawnia się dopiero od pewnego poziomu natężenia treningu.
18
Wrażliwość na insulinę rośnie gwałtownie nawet po jednorazowym treningu fizycznym,
ale szybko wraca do stanu wyjściowego, jeżeli trening nie jest powtarzany. Podobnie u zwierząt, u których ekspozycja na zimno spowodowała dreszcze (skurcze mięśni szkieletowych
spowodowane zimnem) obserwowano poprawę tolerancji glukozy. Również regularnie powtarzanemu treningowi fizycznemu towarzyszy wzrost wrażliwości na insulinę ale 6–10 dni
po przerwaniu treningu jego korzystny efekt zanika i następuje wzrost poziomów glukozy
i insuliny we krwi. Co więcej, wykazano, że u zdrowych osób, które przez 3 doby pozostawały
w łóżku, dochodziło do spadku tolerancji glukozy i wzrostu glikemii i insulinemii. Te i inne
obserwacje wyraźnie dowodzą, że wysiłek fizyczny jest ważnym regulatorem ustrojowego
metabolizmu glukozy również u współczesnego człowieka.
Ponadto wykazano w grupach zdrowych otyłych osób, otyłych osób w podeszłym wieku
i otyłych kobiet z chorobą wieńcową, że trening fizyczny zwiększał ich tolerancję glukozy
niezależnie od wpływu na wagę ciała. Wysiłek fizyczny poprawiał także tolerancję glukozy
u osób, u których była ona obniżona w wyniku przewlekłego głodzenia lub diety niskokalorycznej. Ponadto, wykazano, że dobrze wytrenowani sportowcy mają podobnie podwyższoną
zawartość triglicerydów w mięśniach szkieletowych jak otyli cukrzycy, a mimo to cechuje
ich wysoka wrażliwość na insulinę. Przytoczone obserwacje dowodzą, że wysiłek fizyczny
korzystnie wpływa na przemiany glukozy w mechanizmie, który jest w znacznej mierze niezależny od faktu, że wysiłkowi towarzyszy poprawa bilansu energetycznego ustroju, spadek
wagi ciała, i ewentualnie mniejsze gromadzenie trójglicerydów w komórkach mięśni szkieletowych.
Wysiłek fizyczny, obok efektów regulacyjnych, dotyczących metabolizmu glukozy i lipidów, ma prawdopodobnie wiele innych korzystnych działań, które w sumie składają się na
wielokrotnie opisywane pro-zdrowotne działanie ruchu. Klasyczne w tym kontekście jest badanie Harward Nurses’ Health Study, w którym badano efekty zdrowotne codziennej aktywności ruchowej w grupie zdrowych pielęgniarek, i na wynikach którego opierają się amerykańskie zalecenia zdrowotne dotyczące aktywności fizycznej. Prospektywna obserwacja wykazała istnienie dramatycznych różnic zdrowotnych między grupą nieaktywnych i aktywnych
fizycznie pielęgniarek. Różnicującym kryterium aktywności fizycznej był fakt codziennego
wykonywania (lub niewykonywania) wysiłku fizycznego, któremu towarzyszy wydatek energetyczny potrzebny na odbycie szybkiego spaceru trwającego 21 minut (~100 kJ/dzień lub
2,5 godziny wysiłku/tygdzień). Otóż okazało się, że u tak zdefiniowanych osób nieaktywnych
fizycznie częściej występowały: udar mózgu (o 117%), cukrzyca typu II (o 85%), choroba
wieńcowa (o 43%), kamica żółciowa (o 49%), rak jelita grubego (o 85%), rak sutka (o 22%)
i zgony (o 41%). W opinii F.W. Booth’a, propagatora hipotezy dotyczącej „genów aktywności
ruchowej”, wyniki te sugerują, że „umiarkowany wysiłek fizyczny, jaki towarzyszy 30-mintowemu szybkiemu spacerowi dziennie, zapewnia organizację i równowagę ekspresji genów
w stopniu tak bliskim poziomowi występującemu u naszych paleolitycznych przodków, że
powoduje to około 30% redukcję ryzyka występowania wielu chorób cywilizacyjnych.”. Autor
ten równocześnie zauważa, że około 70% populacji USA nie wykonuje rekomendowanych
30 minut codziennych ćwiczeń fizycznych.
Wraz z rozwojem cywilizacyjnym życie człowieka coraz bardziej różni się od wzorca
z epoki kamiennej. Dwie różnice wydają się szczególnie ważne:
19
1. Wobec swobodnego dostępu do pokarmu, nastąpiło przerwanie naturalnego cyklu okresów sytości i głodu (ryc. 1.7.). Skutkuje to nadmiernym magazynowaniem substratów energetycznych i ewentualną nadwagą i otyłością. Winowajcą mogą być „gospodarne geny”
wymuszające niepotrzebnie oszczędne gospodarowanie zasobami energetycznymi ustroju. Inny powód to dodatni bilans energetyczny ustroju. Choć spożywamy obecnie mniej
kalorii niż nasi przodkowie, to nieporównanie mniej niż oni ich spalamy ze względu na
siedzący tryb życia.
2. Nastąpiła całkowita utrata związku między zdobywaniem pokarmu i wysiłkiem fizycznym. Zakładając, że wysiłek fizyczny jest genetycznie uwarunkowanym regulatorem ważnych procesów
metabolicznych, jego brak staje się czynnikiem zaburzającym homeostazę ustroju w sposób niezależny od wpływu na wagę ciała. Za koncepcją tą przemawiają obserwacje, że trening fizyczny
ma działanie prozdrowotne nawet wtedy, kiedy nie powoduje spadku wagi ciała.
Przynajmniej cztery grupy argumentów przemawiają na korzyść teorii, że zespół kardio-metaboliczny powstaje w wyniku kolizji archaicznego genomu z nowożytnymi warunkami życia:
1. Badania genetyczne wykazały, że na przestrzeni ostatnich 10 tysięcy lat nie było istotnych
zmian w strukturze ludzkiego DNA;
2. Fakt, że epidemia zespołu metabolicznego jest kwestią ostatnich dziesięcioleci daje się tłumaczyć urbanizacją, postępem mechanizacji życia i tym, że życie codzienne i produkcja
różnych dóbr coraz rzadziej wymagają wysiłku fizycznego. Dodatkowo w zachodnich społeczeństwach praktycznie nie występują okresy głodu, tak częste jeszcze na przełomie dziewiętnastego i dwudziestego wieku (np. częsty głód na wsiach na przednówku);
3. Podobną argumentację można zastosować przy interpretacji faktu, że nasilenie zespołu
kardio-metabolicznego u imigrantów z krajów trzeciego świata zamieszkałych w krajach
zachodnich jest znacznie większe niż u ich krewnych żyjących w krajach pochodzenia (epidemia zespołu kardio-metabolicznego wśród meksykańskich imigrantów w USA);
4. Ważkie argumenty na rzecz dyskutowanej hipotezy pochodzą z badań porównawczych dotyczących współczesnych populacji myśliwych-zbieraczy. Klasyczne jest tu badanie amerykańskich Indian Pima zamieszkujących zarówno w Meksyku jak i w Arizonie w USA [16].
Grupy te rozdzieliły się około 700–1000 lat temu, więc jest bardzo prawdopodobne, że ich
genotypy są ciągle niemal identyczne. Indianie meksykańscy prowadzą ciągle tradycyjny
rolniczo-zbieraczo-myśliwski tryb życia, żywią się dietą roślinną i około 20 godzin w tygodniu zajmuje im praca fizyczna. Natomiast Indianie arizońscy żyją w rezerwatach i prowadzą typowy amerykański, siedzący i dostatni tryb życia, ich dieta składa się w 40% z tłuszczy,
a praca fizyczna zajmuje im mniej niż 5 godzin tygodniowo. Okazuje się, że w porównaniu
z Indianami meksykańskimi, ich arizońscy krewni są przeciętnie o 26 kg ciężsi i około sześć
razy częściej mają cukrzycę (połowy z nich zapada na cukrzycę przed 35-tym rokiem życia).
Podobne obserwacje przyniosły badania porównawcze dotyczące innych populacji współczesnych zbieraczy-myśliwych (Eskimosi, mieszkańcy Nowej Gwinei).
Teoria ewolucyjna, choć atrakcyjna, ciągle wymaga weryfikacji. Natomiast już dzisiaj dostępne są dane pokazujące, że zarówno otyłość jak i brak ruchu (per se lub z udziałem starożytnego genomu) mogą prowadzić do zaburzeń metabolicznych skutkujących zaburzeniami
w układzie sercowo-naczyniowym (rozdz. X) będących prawdopodobną przyczyną pogarszania się współczesnego trendu umieralności sercowo-naczyniowej (ryc. 1.5.).
20
I.6. Historia naturalna miażdżycy
Całkowita powierzchnia zmian
(mm2/sekcję)
Odsetek aort ze zmianami
(%)
Obecna wiedza na temat historii naturalnej miażdżycy u ludzi pochodzi z badań autopsyjnych i zawdzięczamy ją głównie jednej grupie badawczej włoskiej i dwóm amerykańskim [17].
Płodowe pochodzenie miażdżycy; patolodzy włoscy przebadali [18] 82 aorty pochodzące od spontanicznie poronionych płodów oraz wcześniaków zmarłych tuż po urodzeniu
(wiek płodowy 6,2 ± 1,3 miesięcy) [19]. Część matek badanych płodów/wcześniaków miała
hipercholesterolemię przed ciążą i w czasie ciąży, część jedynie w ciąży, a część miała prawidłowy poziom cholesterolu. Poziom cholesterolu u płodów nie korelował jednak z jego poziomem u matek. Aorty dzielono na: łuk aorty, aortę piersiową i brzuszną, i z każdego segmentu
uzyskiwano 90 kolejnych poprzecznych sekcji histologicznych, poddawanych następnie badaniu histologicznemu i immunohistochemicznemu. Zmiany miażdżycowe, w postaci pasm
tłuszczowych (fatty streaks), stwierdzono w 60–80% badanych aort, niezależnie od tego czy
pochodziły one od płodów/wcześniaków matek z normalnym czy podwyższonym cholesterolem. Natomiast zaawansowanie zmian mierzone ich całkowitą powierzchnią rosło proporcjonalnie do stopnia zaawansowania hipercholesterolemii u matek (ryc. 1.8.).
W zmianach sklasyfikowanych jako miażdżycowe, występowało pogrubienie intimy oraz
gromadzenie się: makrofagów, natywnych i oksydowanych LDL, komórek piankowatych
i pozakomórkowych skupisk lipidów. W istocie, skład komórkowy tych zmian był podobny
do znajdowanego w pasmach tłuszczowych w tętnicach osobników dorosłych. Podobnie jak
u dorosłych, zmiany były najczęściej obecne w aorcie brzusznej, a najrzadziej w piersiowej.
Dane te sugerują, że zmiany u płodów/wcześniaków były typowymi zmianami miażdżycowymi, i że miażdżyca ma często swój początek już w życiu płodowym.
Ryc. 1.8. Zmiany miażdżycowe w aortach 82 przedwcześnie zmarłych płodów ludzkich pochodzących
od matek z normocholesterolemią (n = 22), hipercholesterolemią występującą jedynie w czasie ciąży
(n = 33) oraz hipercholesterolemią obecną przed i w czasie ciąży (n = 27). A – procent aort, w których
występowały wczesne zmiany miażdżycowe; B – średnia powierzchnia zmian miażdżycowych Wykres
na podstawie danych z referencji [19].
21
Całkowita powierzchnia zmian
(mm2/sekcję)
Miażdżyca jest chorobą całej populacji zachodniej bez względu na wiek; badacze włoscy przebadali, w opisany powyżej sposób, także dzieci w wieku od 1–13 roku życia, które
zginęły w wyniku wypadków lub zmarły z powodu tętniaka tętnicy mózgowej lub nowotworu [20]. U wszystkich tych osobników obecne były zmiany miażdżycowe w aorcie brzusznej
i łuku aorty, mimo że u wszystkich poziom cholesterolu był normalny. U młodszych dzieci były to głównie pasma tłuszczowe, a u osobników powyżej 10-go roku życia stwierdzano
dodatkowo pogrubienie intimy i częste występowanie klasycznych blaszek miażdżycowych.
Zaawansowanie zmian miażdżycowych, mierzone całkowitą ich powierzchnią, rosło liniowo
wraz z wiekiem, ale szybkość narastania zmian była znamiennie większa u dzieci matek z hipercholesterolemią (ryc. 1.9.). Sugeruje to, że ekspozycja na zwiększony poziom cholesterolu
w życiu płodowym pozostawia długotrwałą „pamięć” skutkującą szybszym rozwojem miażdżycy w życiu pozapłodowym [18].
Ryc. 1.9. Rozmiar zmian miażdżycowych w aorcie brzusznej dzieci rośnie wraz z ich wiekiem i wzrost
ten jest znacznie szybszy u dzieci matek z hipercholesterolemią (V) w porównaniu z dziećmi matek
z normocholesterolemią (Ο). Wykres na podstawie danych z referencji [20].
W tym kontekście autorzy spekulują, że jest to konsekwencją, znanego w biologii rozwojowej
procesu znanego jako „programowanie wewnątrzmaciczne” (in-utero programming) lub „programowanie płodowe” (fetal programming). Innym przykładem tego zjawiska, może być korelacja
pomiędzy niską wagą urodzeniową ludzkich noworodków i późniejszym występowaniem u tych
osobników zespołu kardio-metabolicznego i różnych schorzeń miażdżyco-pochodnych [21].
The Bogalusa Heart Study jest badaniem epidemiologicznym dotyczącym wielorasowej
(65% biali, 35% Murzyni) społeczności miejskiej w miasteczku Bogalusa w stanie Luizjana,
USA. W ramach tego badania, patolodzy z Uniwersytetu w Nowym Orleanie, opublikowali wyniki autopsji 204 osób w wieku 2–39 lat, które zmarły w wyniku wypadku, zabójstwa
lub samobójstwa [22]. U wszystkich badanych stwierdzono pasma tłuszczowe w aorcie. Natomiast w tętnicach wieńcowych pasma tłuszczowe miało ~50% osobników w wieku 2–15
i ~85% w wieku 21–39 lat. Rozwinięte blaszki miażdżycowe w tętnicach wieńcowych miało
10% osób w wieku 2–15 lat i 60% osób w wieku 26–39 lat. Badanie wykazało związek między
standardowymi czynnikami ryzyka (palenie papierosów, nadciśnienie, hipercholesterolemia
etc.) a liczbą i rozległością zmian miażdżycowych.
22
Odsetek GPZ ze stenozą
(%)
Odsetek GPZ ze zmianami
(%)
Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Reaserch Group bada
młodych ludzi w przedziale wieku 15–34 lat. W badaniu bierze udział 9 zakładów patomorfologii z całych Stanów Zjednoczonych, co pozwala przyjąć, że badania „Grupy PDAY” są
reprezentatywne dla całej populacji USA. W ramach tego badania opublikowano dane dotyczące zmian miażdżycowych u 1532 osób, które zmarły w sposób nagły (wypadek, zabójstwo,
samobójstwo) [23]. Wszyscy badani osobnicy mieli zmiany miażdżycowe w aorcie a częstość
występowania zmian w tętnicach wieńcowych wynosiła około 50% w grupie wiekowej 15–19
lat i rosła gwałtownie w starszych grupach. W aorcie i tętnicach wieńcowych wraz z wiekiem
rosły także rozmiary zmian i stopień ich zawansowania. Rozmiary i stopień zaawansowania zmian był większy u mężczyzn niż u kobiet i był podobny u białych i czarnych osobników. Pouczającą statystykę opublikowaną przez „Grupę PDAY”, a dotyczącą występowania
zmian miażdżycowych tylko w jednej tętnicy wieńcowej (GPZ) [24], pokazuje ryc. 1.10. Grupa PDAY opublikowała kilkanaście analiz dowodzących [24-31], że różne czynniki ryzyka
zwłaszcza, jeżeli występują równocześnie, zwiększają częstość i stopień zaawansowania zmian
miażdżycowych w tętnicach wieńcowych [24,27,28,32].
Ryc. 1.10. Występowanie zmian miażdżycowych w gałęzi przedniej zstępującej l. tętnicy wieńcowej
(GPZ) u 760 Amerykanów w wieku 15–34 lat zmarłych z powodu wypadku, zabójstwa lub samobójstwa.
Wraz z wiekiem rosła częstość występowania wszystkich rodzajów zmian miażdżycowych (góra) a także
częstość występowania blaszek powodujących istotną stenozę wieńcową (dół). Można przypuszczać, że
gdyby zbadano pozostałe tętnice wieńcowe to częstość występowania zmian miażdżycowych w tętnicach wieńcowych badanych osobników byłaby bliska 100%. Dane z referencji [24].
23
Podsumowując, obecna wiedza o historii naturalnej miażdżycy pochodzi głównie z badań autopsyjnych wykonanych w USA i Włoszech. Brak analogicznych badań spoza kręgu
cywilizacji zachodniej. Wiemy obecnie, że zmiany miażdżycowe w ludzkiej aorcie: (i) powstają nawet w życiu płodowym; (ii) występują praktycznie w całej populacji krajów cywilizacji
zachodniej; (iii) wykazują szybką progresję wraz z wiekiem; (iv) o powstawaniu i dynamice
rozwoju zmian miażdżycowych decyduje obecność klasycznych czynników ryzyka choroby
sercowo-naczyniowej oraz (v) wrażliwość na pro-miażdżycowe działanie cholesterolu, i być
może innych czynników ryzyka, jest programowana już w trakcie życia płodowego. Badania
dotyczące miażdżycy tętnic wieńcowych są mniej kompletne, ale dowodzą, że są one mniej
wrażliwe na czynniki aterogenne niż aorta brzuszna.
Piśmiennictwo
1. Gersh BJ, Sliwa K, Mayosi BM, Yusuf S. Novel therapeutic concepts: the epidemic of cardiovascular
disease in the developing world: global implications. Eur Heart J 2010; 31:642-648.
2. Fuster V, Moreno PR. Atherothrombosis as a systemic, often silent, disease. Nat Clin Pract Cardiovasc Med 2005; 2:431.
3. Aronow WS, Ahn C. Prevalence of coexistence of coronary artery disease, peripheral arterial disease,
and atherothrombotic brain infarction in men and women > or = 62 years of age. Am J Cardiol 1994;
74:64-65.
4. Criqui MH, Denenberg JO. The generalized nature of atherosclerosis: how peripheral arterial disease
may predict adverse events from coronary artery disease. Vasc Med 1998; 3:241-245.
5. Lee AJ, Price JF, Russell MJ, Smith FB, van Wijk MC, Fowkes FG. Improved prediction of fatal
myocardial infarction using the ankle brachial index in addition to conventional risk factors: the
Edinburgh Artery Study. Circulation 2004; 110:3075-3080.
6. Ford ES, Capewell S. Proportion of the decline in cardiovascular mortality disease due to prevention
versus treatment: public health versus clinical care. Annu Rev Public Health 2011; 32:5-22.:5-22.
7. Ford ES, Capewell S. Coronary heart disease mortality among young adults in the U.S. from 1980
through 2002: concealed leveling of mortality rates. J Am Coll Cardiol 2007; 50:2128-2132.
8. Nemetz PN, Roger VL, Ransom JE, Bailey KR, Edwards WD, Leibson CL. Recent trends in the
prevalence of coronary disease: a population-based autopsy study of nonnatural deaths. Arch Intern
Med 2008; 168:264-270.
9. Rosengren A, Dotevall A, Eriksson H, Wilhelmsen L. Optimal risk factors in the population: prognosis,
prevalence, and secular trends. Data from Göteborg population studies. Eur Heart J 2001; 22:136-144.
10. Van Horn L, Greenland P. Prevention of coronary artery disease is a pediatric problem. JAMA 1997;
278:1779-1780.
11. Neel JV. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum
Genet 1962; 14:353-362.
12. Neel JV. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? 1962. Bull World
Health Organ 1999; 77:694-703.
13. Booth FW, Chakravarthy MV, Gordon SE, Spangenburg EE. Waging war on physical inactivity: using modern molecular ammunition against an ancient enemy. J Appl Physiol 2002; 93:3-30.
14. Chakravarthy MV, Booth FW. Eating, exercise, and “thrifty” genotypes: connecting the dots toward
an evolutionary understanding of modern chronic diseases. J Appl Physiol 2004; 96:3-10.
15. Cordain L, Gotshall RW, Eaton SB, Eaton SB, III. Physical activity, energy expenditure and fitness: an
evolutionary perspective. Int J Sports Med 1998; 19:328-335.
24
16. Knowler WC, Pettitt DJ, Bennett PH, Williams RC. Diabetes mellitus in the Pima Indians: genetic
and evolutionary considerations. Am J Phys Anthropol 1983; 62:107-114.
17. Beręsewicz A, Skierczyńska A. Miażdżyca – choroba całego życia i całej populacji krajów cywilizacji
zachodniej. Choroby Serca i Naczyń 2006; 3:1-6.
18. Palinski W, Napoli C. The fetal origins of atherosclerosis: maternal hypercholesterolemia, and cholesterol-lowering or antioxidant treatment during pregnancy influence in utero programming and
postnatal susceptibility to atherogenesis. FASEB J 2002; 16:1348-1360.
19. Napoli C, D’Armiento FP, Mancini FP, Postiglione A, Witztum JL, Palumbo G, Palinski W. Fatty
streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J Clin Invest 1997; 100:2680-2690.
20. Napoli C, Glass CK, Witztum JL, Deutsch R, D’Armiento FP, Palinski W. Influence of maternal hypercholesterolaemia during pregnancy on progression of early atherosclerotic lesions in childhood:
Fate of Early Lesions in Children (FELIC) study. Lancet 1999; 354:1234-1241.
21. Barker DJ, Clark PM. Fetal undernutrition and disease in later life. Rev Reprod 1997; 2:105-112.
22. Berenson GS, Srinivasan SR, Bao W, Newman WP, III, Tracy RE, Wattigney WA. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults. The
Bogalusa Heart Study. N Engl J Med 1998; 338:1650-1656.
23. Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. Natural history
of aortic and coronary atherosclerotic lesions in youth. Findings from the PDAY Study. Arterioscler
Thromb 1993; 13:1291-1298.
24. McGill HC, Jr., Mcmahan CA, Zieske AW, Tracy RE, Malcom GT, Herderick EE, Strong JP. Association of coronary heart disease risk factors with microscopic qualities of coronary atherosclerosis in
youth. Circulation 2000; 102:374-379.
25. McGill HC, Jr., Mcmahan CA, Herderick EE, Zieske AW, Malcom GT, Tracy RE, Strong JP. Obesity accelerates the progression of coronary atherosclerosis in young men. Circulation 2002; 105:2712-2718.
26. Strong JP, Malcom GT, Oalmann MC, Wissler RW. The PDAY Study: natural history, risk factors, and
pathobiology. Pathobiological Determinants of Atherosclerosis in Youth. Ann N Y Acad Sci 1997;
811:226-235 (discussion 235-7).
27. Zieske AW, Malcom GT, Strong JP. Natural history and risk factors of atherosclerosis in children and
youth: the PDAY study. Pediatr Pathol Mol Med 2002; 21:213-237.
28. Zieske AW, Mcmahan CA, McGill HC, Jr., Homma S, Takei H, Malcom GT, Tracy RE, Strong JP.
Smoking is associated with advanced coronary atherosclerosis in youth. Atherosclerosis 2005;
180:87-92.
29. McGill HC, Jr., Mcmahan CA, Tracy RE, Oalmann MC, Cornhill JF, Herderick EE, Strong JP. Relation of a postmortem renal index of hypertension to atherosclerosis and coronary artery size in
young men and women. Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. Arterioscler Thromb Vasc Biol 1998; 18:1108-1118.
30. McGill HC, Jr., Mcmahan CA, Malcom GT, Oalmann MC, Strong JP. Effects of serum lipoproteins
and smoking on atherosclerosis in young men and women. The PDAY Research Group. Pathobiological Determinants of Atherosclerosis in Youth. Arterioscler Thromb Vasc Biol 1997; 17:95-106.
31. McGill HC, Jr., Mcmahan CA, Malcom GT, Oalmann MC, Strong JP. Relation of glycohemoglobin
and adiposity to atherosclerosis in youth. Pathobiological Determinants of Atherosclerosis in Youth
(PDAY) Research Group. Arterioscler Thromb Vasc Biol 1995; 15:431-440.
32. Zieske AW, Tracy RP, Mcmahan CA, Herderick EE, Homma S, Malcom GT, McGill HC, Jr., Strong
JP. Elevated serum C-reactive protein levels and advanced atherosclerosis in youth. Arterioscler
Thromb Vasc Biol 2005; 25:1237-1243.
25
II. Energetyka serca
Andrzej BERĘSEWICZ
II.1. Podstawowe pojęcia i liczby
Serce dorosłego człowieka kurczy się około 100 000 razy i pompuje 6–7 tysięcy litrów
krwi na dobę (tab. 2.1.). Bezpośrednim źródłem energii dla tej ogromnej pracy mechanicznej
pompy sercowej są wysokoenergetyczne wiązania reszt kwasu fosforowego w cząsteczkach
adenozynotrifosforanu (ATP).
Tab. 2.1. Serce dorosłego człowieka w liczbach
Waga serca
Liczba kardiomiocytów
Liczba cykli
Rzut skurczowy
Rzut minutowy
Rzut dobowy
Produkcja ATP
Praca hemodynamiczna
Zapas ATP w komórce
Efektywność energetyczna pompy sercowej
Rezerwa rzutu minutowego
Rezerwa wieńcowa
Rezerwa tlenowej produkcji ATP
250 g.
5–6 mld
100 000/dobę
70 ml
5 litrów/min
6–7 tys. l/dobę
3–5 kg/dobę
zużywa 80% produkcji ATP
na 10 sek.
20%
3–4
3–4
5–6
Serce produkuje 3–5 kg ATP/dobę, z czego ~80% zasila proces skurczu i rozkurczu mięśnia sercowego. Pozostałe 20% zasila czynność elektryczną, transporty jonowe oraz podstawowe szlaki metaboliczne w sercu.
Ogromną i trwającą przez całe życie sercową produkcję ATP zapewniają w ponad 95%
tlenowe przemiany niezestryfikowanych kwasów tłuszczowych (NEFA), glukozy i kwasu
mlekowego zachodzące w mitochondriach (proces określany jako spalanie substratów energetycznych). Źródłem pozostałych ~5% ATP jest glikoliza – proces niewymagający udziału
27
tlenu. Przemiany te utrzymują komórkowe stężenia ATP i fosfokreatyny (CrP) w sercu na
względnie stałym poziomie, nawet w czasie 3–4-krotnego wzrostu rzutu minutowego serca,
jaki może towarzyszyć wysiłkowi fizycznemu u młodych zdrowych osób.
Serce jest narządem wyspecjalizowanym w tlenowej produkcji ATP. Wskazuje na to fakt,
że mitochondria zajmują około 1/3 objętości kardiomiocytów, naczynia mikrokrążenia około
1/3 objętości tkanki sercowej, a gęstość naczyń włosowatych w sercu jest 2–3-krotnie większa
niż w mięśniu szkieletowym i 10-krotnie większa niż w mózgu, w efekcie, każdy kardiomiocyt
kontaktuje się z kilkoma włośniczkami. Serce, w porównaniu z mózgiem, który jest również
bardzo energochłonnym narządem, ma znacznie większą perfuzję i konsumpcję tlenu (O2) na
gram tkanki i znacznie większą ekstrakcję O2. Serce mimo że stanowi tylko 0,4% wagi ciała
zużywa aż 11% ustrojowej konsumpcji O2 (tab. 2.2.).
Tabela 2.2. Specjalizacja serca w tlenowej produkcji ATP
Serce
Mózg
Naczynia mikrokrążenia
Gęstość naczyń włosowatych
Mitochondria (% objętości komórek)
40% objętości tkanki
3–4 tys./mm2
30%
340/mm2
<10%
Waga
Waga (% wagi ciała)
250 g
0,4%
1400 g
2%
Perfuzja na g. tkanki
Całkowita perfuzja narządowa
Perfuzja w % rzutu minutowego
1 ml/min/g
250 ml/min
4%
0,33 ml/min/g
750 ml/min
13%
Ekstrakcja O2
Konsumpcja O2
% ogólnoustrojowej konsumpcji O2
70%
0,1 ml/min/g
11%
35%
0,06 ml/min/g
20%
Następujące elementy składają się na specyfikę energetyki mięśnia sercowego w porównaniu z energetyką innych narządów [1-3]:
1. Komórkowe zapasy ATP (5 μmoli/g mokrej wagi) i CrP (6 μmoli/g mokrej wagi) w sercu są
niewielkie i wystarczają jedynie na kilkanaście kolejnych skurczów [2];
2. Nawet w spoczynku, minutowa produkcja i rozkład ATP (~30 μmoli/g mokrej wagi/min)
są wielokrotnie większe od zapasowego ATP [2];
3. Dla utrzymania prawidłowej czynności skurczowej serca (zwłaszcza w czasie wysiłku fizycznego czy stymulacji współczulnej, kiedy rzut minutowy może rosnąć nawet 3–4-krotnie),
jest niezbędne by produkcja ATP w mitochondriach i jego transport z mitochondriów do
aparatu kurczliwego (rozdz. II.2) ściśle nadążały za zużyciem ATP. W spoczynku mitochondria wykorzystują jedynie 15–20% swojego maksymalnego potencjału tlenowej produkcji
ATP. Oznacza to, że serce dysponuje 5–6-krotną rezerwą tlenowej produkcji ATP [2];
4. Nienadążanie sercowej produkcji/transportu ATP za obciążeniem skutkuje utratą kurczliwości mięśnia sercowego już w czasie kilku kolejnych cykli sercowych. Najczęstszym powodem tego deficytu (oraz zaburzeń kurczliwości i dolegliwości wieńcowych) jest upośle28
dzenie produkcji ATP związane z niedokrwieniem. Inną przyczyną może być „trwonienie”
ATP w procesach niezwiązanych ze skurczem, co skutkuje obniżeniem tzw. sprawności mechanicznej pompy sercowej (rozdz. II.2) (zjawisko typowe dla reperfundowanego i ogłuszonego, a także dla niewydolnego mięśnia sercowego);
5. Tlenowy metabolizm dostarcza >95% sercowego ATP, dlatego skurcz mięśnia sercowego
w sposób krytyczny zależy od dostępności O2, a ta od wielkości perfuzji wieńcowej. Już
w spoczynku, miokardium pobiera z przepływającej przez nie krwi prawie całą możliwą do
pozyskania ilość O2 (ekstrakcja O2 w sercu wynosi 65–70%, wobec ~35% w mózgu i mięśniach szkieletowych, Tab. 2) (ryc. 2.1.). Dlatego, kiedy rośnie obciążenie i zapotrzebowanie
serca na O2, wzrost przepływu wieńcowego jest jedynym sposobem zwiększania podaży O2
i utrzymania równowagi energetycznej serca. Podobnie, nawet niewielki spadek perfuzji
wieńcowej skutkuje deficytem ATP, ponieważ nie może być kompensowany większą ekstrakcją O2. Zapoczątkowuje to błędne koło regulacyjne skutkujące dalszym pogorszeniem
bilansu energetyczny (deficyt ATP skutkuje zaburzeniami kurczliwości i hipotonią, która
jeszcze bardziej upośledza perfuzję miokardium);
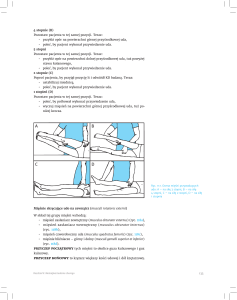
Ryc. 2.1. Krzywa dysocjacji hemoglobiny (Hb). Ciśnienie parcjalne tlenu (pO2) we krwi tętniczej wynosi ~100 mmHg, przy którym saturacji hemoglobiny O2 wynosi ~100% (punkt A). W większości tkanek:
pO2 krwi żylnej wynosi ~40 mmHg, saturacja Hb ~75% (punkt B), ekstrakcja O2 ~25% i w razie potrzeby ekstrakcja może wzrastać nawet trzykrotnie (punkt D). W sercu w spoczynku pO2 w zatoce wieńcowej wynosi ~22–25 mmHg, saturacja Hb ~35–38% (punkt C), a ekstrakcja O2 ~65%, co jest wartością
bliską maksymalnej. W wysiłku ekstrakcja może wzrosnąć zaledwie o kilka procent (punkt D).
6. W normalnym sercu przepływ wieńcowy ściśle dostosowuje się do aktualnego obciążenia
serca (w literaturze angielskiej zjawisko znane jako perfusion-contraction matching) [4] i nie
ogranicza możliwości wykonywania wysiłku. Świadczy o tym fakt, że u zdrowych osobników, nawet podczas maksymalnego wysiłku, krążenie wieńcowe ma jeszcze pewną rezerwę
wazodylatacyjną [5,6] (ryc. 2.2). Stosunek przepływu w czasie maksymalnego rozkurczu
naczyń wieńcowych pod wpływem substancji rozszerzającej naczynia (np. adenozyna lub
dipirydamol) do spoczynkowego przepływu wieńcowego określany jest terminem „rezerwa wieńcowa” i wynosi w zdrowym sercu 3–4. Mechanizm dostosowywania się przepływu
do obciążenia jest zaburzony w miażdżycy tętnic wieńcowych (rozdz. III), co jest głównym
elementem patomechanizmu choroby wieńcowej;
29
Ryc. 2.2. Liniowa zależność między obciążeniem serca i wielkością przepływu wieńcowego. W normie,
w czasie maksymalnego wysiłku fizycznego ciągle obecna jest ~20% rezerwa wazodilatacyjna krążenia
wieńcowego, co oznacza, że przepływ wieńcowy nie jest czynnikiem ograniczającym zdolność wykonywania wysiłku
7. Ponad 95% sercowego ATP powstaje na bieżąco w wyniku tlenowych przemian substratów
energetycznych w mitochondriach i około 80% tak powstałego ATP zasila skurcz mięśnia
sercowego. Redukcja jednej cząsteczki O2 w procesie fosforylacji oksydacyjnej w mitochondriach skutkuje, w optymalnych warunkach, syntezą ~6 cząsteczek ATP. Dlatego z dużą
precyzją można powiedzieć, że o bilansie energetycznym (i stanie kurczliwości) mięśnia
sercowego decyduje stan równowagi między podażą i konsumpcją O2 przez serce (ryc. 2.3.),
i że pomiar konsumpcji tlenu (MVO2) jest dobrym wskaźnikiem wydatku energetycznego
serca (konsumpcji ATP), a tym samym, jego obciążenia (1 ml O2 jest ekwiwalentem 20 J
energii).
Ryc. 2.3. Czynniki wpływające na równowagę między podażą (lewa strona) i zapotrzebowaniem/konsumpcją tlenu przez serce. Czynniki zwiększające rytm, obciążenie następcze (którego miarą jest napięcie w ścianie) i/lub kurczliwość serca zwiększają konsumpcję tlenu i równocześnie zmniejszają jego
efektywność mechaniczną (efektywność = praca wyrzutu/MVO2). Dzieje się tak dlatego, gdyż w normalnym sercu jedynie ~20% MVO2 zasila wyrzut.
30
II.2. Zużycie energii, konsumpcja O2 i efektywność mechaniczna serca
W pompach mechanicznych jedynie część zużywanej energii jest przekształcana w zewnętrzną pracę mechaniczną i wielkość ta określana jest jako efektywność mechaniczna. Pracą
zewnętrzną w sercu jest praca związana z wyrzutem krwi do aorty i tętnicy płucnej. Efektywność mechaniczna pompy sercowej wynosi około 20% (efektywność = zewnętrzna praca serca/
konsumpcja ATP lub zewnętrzna praca/MVO2), co oznacza, że jedynie ~20% całkowitej puli
ATP lub O2 konsumowanego przez serce zasila pracę związaną z wyrzutem. Pozostała część
zasila tzw. pracę wewnętrzną serca i energia na nią wydatkowana jest tracona w postaci ciepła.
Czynniki, które zwiększają obciążenie następcze komór, którego miarą w przypadku lewej komory jest napięcie w jej ścianie w momencie otwierania się zastawki aortalnej (napięcie
ściany komory, τ = ½ średnicy lewej komory pod koniec rozkurczu x ciśnienie w momencie
otwierania się zastawki aortalnej), zmniejszają efektywność mechaniczną serca, ponieważ
skutkują wzrostem energochłonnej pracy wewnętrznej (związanej ze skurczem izowolumetrycznym komór i generacją ciśnienia), kosztem ograniczenia zakresu skracania komór w fazie wyrzutu i tym samym, pracy wyrzutu. Także czynniki zwiększające kurczliwość i częstość
pracy serca zmniejszają efektywność mechaniczną serca. Dlatego jest ona największa u osobników z wolną czynnością serca, małymi rozmiarami komór, niskim ciśnieniem tętniczym
oraz małą kurczliwością miokardium.
Tab. 2.3. Wzrost MVO2 towarzyszący wzrostowi danej funkcji serca o 50%
Zwiększająca się czynność serca o 50%
Wzrost MVO2
Rytm serca
Praca związana z generacją ciśnienia w komorze
Kurczliwość miokardium
Praca wyrzutu krwi z komory
50%
45%
50%
4%
W warunkach klinicznych, redukcję obciążenia energetycznego serca i redukcję MVO2
najłatwiej uzyskuje się poprzez zwolnienie częstości pracy serca (np. β-blokery, iwabradyna)
i/lub zmniejszenie jego kurczliwości (β-blokery, blokery kanału wapniowego) (tab. 2.3.).
II.3. Podwójny produkt – nieinwazyjny wskaźnik MVO2
Obciążenie serca można pośrednio oszacować poprzez pomiar MVO2. Jednakże bezpośredni pomiar MVO2 wymaga cewnikowania serca i jest rzadko stosowany. Bezpośrednie
pomiary wykazały jednak, że MVO2 w sposób liniowy koreluje z częstością pracy serca, napięciem w ścianie komory (τ = ciśnienie tętnicze x ½ średnicy lewej komory) i kurczliwością
mięśnia sercowego (tab. 2.3.).
Badania eksperymentalne i kliniczne wykazały, że MVO2 szczególnie dobrze koreluje
z tzw. podwójnym produktem (ang. Double Produkt, DP albo Pressure Rate Product), który
oblicza się z równania: DP = częstość serca x ciśnienie tętnicze. Podwójny produkt jest obecnie traktowany jako dość wiarygodny, nieinwazyjny, a zatem użyteczny klinicznie, pośredni
wskaźnik MVO2 i obciążenia serca.
31
II.4. VO2max miarą obciążenia energetycznego organizmu
Organizm człowieka korzysta głównie z tlenowej produkcji ATP, dlatego całkowite pochłanianie O2 przez ustrój (VO2) jest dobrą miarą jego obciążenia energetycznego. Obciążenie wyraża się
w postaci tzw. równoważnika metabolicznego (MET, metabolic equivalent). Jeden MET odpowiada spoczynkowemu pochłanianiu O2 przez siedzącą osobę, co wynosi ~3,5 ml O2/kg/min. W trakcie wysiłku VO2 rośnie wielokrotnie. Dla przykładu, szybki chód stanowi obciążenie 3–5 MET, gra
w tenisa 5–7 MET, a gra w squasha czy bieg z szybkością 10–12 km/godz. – 9 MET (tab. 2.4.).
Maksymalne możliwe do osiągnięcia przez danego człowieka zużycie O2 to tzw. VO2max.
Wartość ta jest w dużej mierze uwarunkowana genetycznie, jest wyższa u mężczyzn, rośnie
w wyniku treningu wytrzymałościowego, a spada wraz z wiekiem, wzrostem masy tkanki tłuszczowej i brakiem aktywności fizycznej. VO2max jest parametrem wskazującym na zdolność
do wykonywania wysiłków tlenowych (wytrzymałościowych). Średnie VO2max u mężczyzny
i kobiety w wieku 35 lat wynosi 44 ml/kg/min i 34 ml/kg/min co odpowiada odpowiednio
12 i 10 MET. Najwyższe zmierzone kiedykolwiek wartości VO2max wynosiły odpowiednio
94 i 77 ml/min/kg i zarejestrowano je u sportowców uprawiających biegi narciarskie.
Droga O2 od powietrza atmosferycznego do zużycia przez tkanki obejmuje: (i) wentylację
płucną; (ii) dyfuzję O2 z powietrza pęcherzykowego do krwi płucnych naczyń włosowatych;
(iii) wiązanie O2 z hemoglobiną; (iv) tłoczenie krwi do tkanek (pojemność minutowa serca);
(v) dyfuzję O2 z krwi do tkanek oraz (vi) zużycie O2 przez tkanki w procesie fosforylacji oksydacyjnej. Każdy z tych procesów może rzutować na ostateczne zużycie O2 tlenu przez organizm,
ale u zdrowego człowieka podstawowym czynnikiem determinującym VO2max jest maksymalna pojemność minutowa serca. Dlatego VO2max jest dobrym wskaźnikiem maksymalnej pojemności minutowej serca, a także miarą upośledzenia rezerwy sercowej i zaawansowania niewydolności serca (VO2max <10 ml/kg/min jest wartością kwalifikującą do przeszczepu serca).
Tabela 2.4. Zależność między poziomem aktywności fizycznej, jej kosztem tlenowym i odpowiadającym im równoważnikiem metabolicznym (MET).
Klasa czynnościowa
(NYHA)
IV
32
Objawy
niewydolności
serca
III
Zdrowy – siedzący
tryb życia
II
Zdrowy wysportowany
I
Stan kliniczny
Koszt tlenowy
ml/kg/min
56,0
52,5
49,0
42,0
38,5
35,0
31,5
28,0
24,5
21,0
17,5
14,0
10,5
7,0
3,5
MET
16
15
14
12
11
10
9
8
7
6
5
4
3
2
1
Badania sercowo-płucne polegają na pomiarze ustrojowego VO2 i produkcji CO2 (VCO2)
podczas wysiłku fizycznego o rosnącym nasileniu, dzięki czemu umożliwiają ocenę tzw. progu
beztlenowego, czyli poziomu wysiłku, przy którym mięśnie zaczynają wykorzystywać przemiany beztlenowe jako dodatkowego źródła energii. U zdrowych niewytrenowanych osób
dzieje się to zwykle przy wysiłku podnoszącym VO2 do poziomu 50–60% VO2max. Dochodzi
wtedy do gromadzenia się kwasu mlekowego we krwi, spadku jej pH i dodatkowego uwalniania CO2 zmagazynowanego w wodorowęglanach krwi. Zwiększone powstawanie CO2 skutkuje zwiększonym jego wydalaniem z organizmu w mechanizmie hiperwentylacji (CO2 we krwi
jest regulatorem ośrodka oddechowego). Zatem miarą progu beztlenowego jest wysiłek, przy
którym wentylacja płucna i/lub VCO2 rosną bardziej niż by to wynikało z aktualnego VO2.
Pomiar progu beztlenowego jest wygodną klinicznie procedurą gdyż nie wymaga obciążania
badanej osoby tak dużymi obciążeniami jak w przypadku pomiaru VO2max. Większość wysiłków dnia codziennego odbywa się poniżej progu beztlenowego. Próg beztlenowy jest dobrą
miarą poziomu wytrenowania danego osobnika.
Piśmiennictwo
1. Ingwall JS, Weiss RG. Is the failing heart energy starved? on using chemical energy to support cardiac
function. Circ Res 2004; 95:135-145.
2. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing
heart. Physiol Rev 2005; 85:1093-1129.
3. Knaapen P, Germans T, Knuuti J, Paulus WJ, Dijkmans PA, Allaart CP, Lammertsma AA, Visser FC.
Myocardial energetics and efficiency: current status of the noninvasive approach. Circulation 2007;
115:918-927.
4. Lee SC, Downey HF. Downregulation of oxygen demand in isoprenaline stimulated canine myocardium. Cardiovasc Res 1993; 27:1542-1550.
5. Heusch G. Heart rate in the pathophysiology of coronary blood flow and myocardial ischaemia: benefit from selective bradycardic agents. Br J Pharmacol 2008; 153:1589-1601.
6. Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiol Rev 2008; 88:10091086.
33
III. Fizjologia krążenia wieńcowego
Michał MĄCZEWSKI
Już w spoczynku ekstrakcja O2 z krwi w sercu jest prawie maksymalna (rozdz. II). Dlatego
kiedy wzrasta zapotrzebowanie miokardium na O2, jedynym sposobem zwiększenia jego podaży do serca jest zwiększenie przepływu wieńcowego. Średni spoczynkowy przepływ wieńcowy w mięśniu lewej komory wynosi ~1 ml/min/g miokardium i jest o kilkanaście procent
większy w warstwie podwsierdziowej niż podnasierdziowej. W prawidłowym sercu, w wyniku działania licznych mechanizmów regulacyjnych, przepływ ten może się zwiększać w czasie
wysiłku o 300–400% (rezerwa wieńcowa = ~3–4) i krążenie wieńcowe nie jest czynnikiem
ograniczającym możliwości wykonywania wysiłku (nawet w czasie maksymalnego wysiłku
krążenie wieńcowe ma jeszcze pewną rezerwę) [1,2].
III.1. Anatomia czynnościowa krążenia wieńcowego
Krążenie wieńcowe tworzą coraz bardziej rozgałęziające się naczynia 4 typów (ryc. 3.1.A):
Tętnice nasierdziowe; od aorty, tuż nad zastawką aortalną, odchodzą lewa (dzieląca się na
gałąź przednią zstępującą, GPZ i gałąź okalającą, GO) i prawa tętnica wieńcowa: trzy główne
tętnice wieńcowe, o średnicy około 3–5 mm. Tętnice te oraz ich kolejne rozgałęzienia (między
innymi gałąź przednia zstępująca, gałąź okalająca) należą do tak zwanych tętnic nasierdziowych, gdyż biegną bezpośrednio pod nasierdziem, na powierzchni mięśnia sercowego. Służą
rozprowadzeniu krwi do różnych obszarów serca, stawiają minimalny opór dla przepływu
i podlegają jedynie słabej regulacji nerwowej;
Tętnice przeszywające; odchodzą od tętnic nasierdziowych w kierunku wsierdzia i tworzą w poszczególnych warstwach ściany komory prostopadłe odgałęzienia, które kilkakrotnie
rozgałęziają na coraz drobniejsze naczynia mikrokrążenia;
Naczynia mikrokrążenia (o średnicy <200 μm), na które składają się kolejno: (i) tętniczki
(mają grubą warstwę mięśniową – stosunek grubości ich ściany do światła wynosi 2:3 (vs. 1:4
w tętnicach nasierdziowych) i są bogato unerwione i stawiają bardzo duży opór; (ii) zwieracze
przedwłośniczkowe (15–30 μm) – o szczególnie pogrubionej warstwie mięśniowej; (iii) meta-ar35
teriole (10–20 μm) – z cienką mięśniówką; (iv) włośniczki (5–10 μm) – zbudowane tylko z jednej
warstwy komórek śródbłonka oraz (v) żyłki (15–200 μm) – ze słabo rozwiniętą mięśniówką;
Żyły – biegną w warstwie podwsierdziowej miokardium, a następnie prostopadle w kierunku nasierdzia, gdzie się łączą w żyły nasierdziowe, których spływ znajduje się w zatoce żylnej.
Opór naczyń żylnych jest minimalny [2].
Ryc. 3.1. Schemat części tętniczej unaczynienia wieńcowego lewej komory serca i dystrybucja przezścienna trzech składowych oporu wieńcowego: (A) – w spoczynku; (B) – w czasie wysiłku; (C) – w spoczynku w obecności istotnego zwężenia tętnicy nasierdziowej oraz (D) – w czasie wysiłku w obecności
zwężenia. Rc – opór czynnościowy (czerwone tło); Rk – opór kompresyjny (zielone tło); RA – opór anatomiczny (niebieskie tło). Opór całkowity = Rc + Rk + RA. Strzałki i ich grubość reprezentują kierunek
i wielkość przepływu krwi. Linie przerywane w B i D pokazują wielkość odpowiednich składowych oporu w okresie poprzedzającym wysiłek (porównaj odpowiednio A i B oraz C i D). Szczegóły w tekście.
III.2. Determinanty fizyczne przepływu wieńcowego
III.2.1. Zależności między oporem ciśnieniem i przepływem
Przepływ cieczy (Q) w dowolnym układzie rurek, w tym w krążeniu wieńcowym, opisuje
równanie Ohma:
Q = ΔP/Rw [1]
ΔP – ciśnienie perfuzyjne, definiowane jako różnica między ciśnieniem na początku i na końcu układu;
Rw – całkowity opór naczyniowy (np. wieńcowy).
W przypadku krążenia wieńcowego, przepływ Q (w ml/min) rośnie proporcjonalnie do
różnicy między ciśnieniem w aorcie i prawym przedsionku i maleje proporcjonalnie do oporu
36
wieńcowego (Rw). Wieńcowe ciśnienie perfuzyjne jest względnie stałe, gdyż ciśnienie aortalne zmienia się w niewielkim zakresie a przedsionkowe jest bliskie zera. Dlatego zasadniczym
regulatorem przepływu wieńcowego są zmiany oporu naczyniowego.
Opór, jaki stawia naczynie płynącej cieczy określa prawo Poiseuille’a:
R = 8 ηl/Π r4 [2]
gdzie: R oznacza opór naczyniowy, η – współczynnik lepkości cieczy, l – długość naczynia,
Π – stałą, a r – promień światła naczynia. Podstawienie wartości R do równania [1] pokazuje,
że przepływ w danym segmencie naczyniowym opisuje równanie:
Q = ΔP x Πr4/8ηl [3]
Oznacza to, że w układzie wieńcowym, w którym długość segmentów naczyniowych, lepkość krwi (przy założeniu, że hematokryt jest stały) i ciśnienie perfuzyjne są względnie stałe,
głównym determinantem przepływu jest promień naczyń w czwartej potędze. Wobec tego,
nawet minimalne zmiany promienia skutkują ogromnymi zmianami oporu naczyniowego
i przepływu. W układzie krążenia, w tym i w układzie wieńcowym, opór (promień) danego
segmentu naczyniowego determinują trzy składowe:
1. opór anatomiczny (RA) – uwarunkowany kalibrem naczyń. Pojedyncze naczynia mikrokrążenia mają duży opór anatomiczny w porównaniu z tętnicami nasierdziowymi (R =
8ηl/Πr4). Jednakże o perfuzji mięśnia sercowego (przepływ na jednostkę masy mięśnia)
decyduje opór sieci naczyniowej jako całości w danym fragmencie mięśnia. Na przykład
gałąź przednia zstępująca lewej tętnicy wieńcowej (GPZ) w sercu świni, w wyniku około
13 kolejnych podziałów, staje się źródłem ~500 tys. drobniejszych naczyń, z których jedynie ~200 ma średnicę >300 μm [3]. W układzie składającym się z szeregowo połączonych
naczyń o różnym kalibrze przepływ jest wypadkową ciśnienia perfuzyjnego i sumy oporów
wszystkich części układu (Q = ΔP/Rc, gdzie Rc = R1 + R2 +...+ Rn). W układzie szeregowym
przepływ (w ml/min) we wszystkich jego częściach jest zawsze taki sam. Jednakże rozgałęzienia tętnic wieńcowych w miokardium mają w znacznej mierze układ równoległy (ryc.
3.1.). Wynikają z tego dwie ważne konsekwencje czynnościowe. Układ równoległy umożliwia regulację przepływu w każdym jego segmencie z osobna. Po drugie, o ile w układzie
szeregowym o przepływie i ciśnieniu decyduje suma oporów poszczególnych składowych
układu (ΔP = Q (R1 + R2 + ... + Rn) to w przypadku układu równoległego ważna jest suma
odwrotności oporów poszczególnych składowych układu:
Q
ΔP = ------------------------------- [4]
1/R1 + 1/R2 + ... + 1/Rn
W sumie oznacza to, że w układzie równoległym, dla uzyskania takiego samego przepływu wystarczy znacznie mniejsze ciśnienie perfuzyjne. Bezpośrednie pomiary wykazały, że
różne warstwy mięśnia sercowego nie różnią się zasadniczo wielkością oporu anatomicznego
(ryc. 3.1.A). Miażdżycowe zwężenia tętnic nasierdziowych są najczęstszą przyczyną wzrostu
oporu anatomicznego w krążeniu wieńcowym (ryc. 3.1.C).
2. opór kompresyjny (RK) – związany z uciskiem wywieranym na naczynia z zewnątrz przez
kurczący się mięsień sercowy i ciśnienie wewnątrzkomorowe. Opór ten jest mały w prawej
komorze, a szczególnie duży w warstwie podwsierdziowej lewej komory (do tego stopnia,
że perfuzja tej warstwy odbywa się głównie w fazie rozkurczu) i maleje w kierunku nasier37
dzia, zwiększa się wraz z naprężeniem komory (σ = Pr/2h, obliczanym z równania Laplace’a),
przyspieszeniem rytmu serca i zaburzeniami rozkurczu lewej komory (w obu ostatnich
sytuacjach maleje udział fazy rozkurczu w cyklu sercowym).
3. opór czynnościowy (RC) – związany z obecnością tzw. tonusu naczyniowego, czyli czynnego napięcia mięśni gładkich budujących ścianę naczyń. O tonusie decyduje w ~80% czynne
napięcie tętniczek o średnicy 10–200 μm, którego wielkość jest wypadkową równocześnie
działających czynników kurczących (głównie regulacja miogenna) i rozkurczających (regulacje metaboliczna i śródbłonkowa).
Średni spoczynkowy przepływ wieńcowy jest o kilkanaście procent większy w warstwie
podwsierdziowej niż podnasierdziowej lewej komory. Wobec takiego samego ciśnienia perfuzyjnego w obu warstwach, oznacza to, że całkowity opór naczyniowy jest nieco mniejszy
w warstwie podwsierdziowej. Jak ilustruje to ryc. 3.1., warstwy ściany lewej komory różnią
się udziałem oporu kompresyjnego i czynnościowego w całkowitym oporze wieńcowym.
W zdrowym sercu, zmianom regulacyjnym podlega głównie opór czynnościowy. Od niego
zależy też wielkość tzw. rezerwy rozkurczowej (wieńcowej) mięśnia sercowego. W prawidłowym sercu rezerwa ta jest największa w warstwie podnasierdziowej lewej komory i maleje
w kierunku wsierdzia [2,4,5].
III.2.2. Dystrybucja oporu wieńcowego i koncepcja fractional flow reserve
Wieńcowe drzewo naczyniowe można potraktować jako układ szeregowo połączonych
oporników. Opór całkowity krążenia wieńcowego jest sumą oporów kolejnych segmentów
naczyniowych, zgodnie z równaniem:
Rcałkowity = Rt.nasierdziowe + Rt.przeszywające + Rmikrokrążenie + Rżyły [5]
Podobnie jak całkowity opór wieńcowy, także cząstkowe opory jego segmentów można
wyznaczyć jedynie pośrednio z przekształcenia równania [1]:
R = ΔP/Q [6]
W praktyce bardzo użyteczną miarą oporu danego segmentu naczyniowego jest spadek
ciśnienia, do jakiego w tym segmencie dochodzi w warunkach stałego przepływu. Na przykład dobrą miarą oporu stawianego przez miażdżycowe zwężenie tętnicy jest różnica między
ciśnieniem zmierzonym przed i za zwężeniem (rozdz. III.8.3). Ponieważ przepływ (Q) jest
identyczny we wszystkich segmentach naczyniowych, opór danego segmentu naczyniowego
jest wprost proporcjonalny do spadku ciśnienia, jaki w nim występuje.
Bezpośrednie pomiary w sercu wykazały (ryc. 3.2.), że spadek ciśnienia jest minimalny
w tętnicach nasierdziowych (>500 μm) i tętnicach przeszywających (500–200 μm). Ciśnienie
gwałtownie spada w mikrokrążeniu, w szczególności w tętniczkach mikrokrążenia (zwanych
przez to tętniczkami oporowymi), a praktycznie nie zmienia się w naczyniach żylnych. W efekcie okazuje się, że w krążeniu wieńcowym około 80% całkowitego oporu wieńcowego rezyduje w mikrokrążeniu, a jedynie 20% w większych naczyniach. Nierównomierny rozkład oporu naczyniowego jest spowodowany głównie faktem, że naczynia mikrokrążenia mają bardzo
duże czynne napięcie mięśniówki gładkiej. Dowodzi tego obserwacja, że pod wpływem bodźca
naczyniorozkurczowego (np. wysiłku fizycznego lub substancji naczyniorozkurczającej, np. dipirydamolu) dochodzi do preferencyjnego rozkurczu tętniczek mikrokrążenia, spadku oporu
naczyniowego głównie w tym obszarze i wzrostu przepływu wieńcowego (ryc. 3.2.) [6,7].
38
Ryc. 3.2. Rozkład ciśnień i oporów w krążeniu wieńcowym. Panel górny: Na osi rzędnych, od lewej do prawej, oznaczono malejącą średnicę naczyń tętniczych, następnie naczynia mikrokrążenia <200 μm (czarną
poziomą grubą linią) a następnie rosnące średnice naczyń żylnych. Około 80% spadku ciśnienia odbywa
się w naczyniach mikrokrążenia. Panel dolny: czarne słupki (Normalne) pokazują, jaki procent całkowitego
oporu wieńcowego rezyduje w warunkach normalnych w danym segmencie naczyniowym. Słupki szare (Dipirydamol) pokazują zmianę oporu wieńcowego po podaniu dipirydamolu. Dipirydamol spowodował sześciokrotny spadek całkowitego oporu wieńcowego. Podczas gdy opór segmentu naczyń tętniczych o średnicy
>170 μm zmalał jedynie o 33%, opór segmentu o średnicy <170 μm zmalał aż 15 krotnie (wg [6]).
Wszystko to sprawia, że w zdrowym krążeniu wieńcowym o przepływie wieńcowym decyduje średnica (opór) naczyń mikrokrążenia, a zmiany w innych segmentach naczyniowych
mają minimalne znaczenia. Tezę te ilustruje fakt, że blaszka miażdżycowa ograniczająca średnicę tętnicy nasierdziowej o 50% i zwiększająca kilkunastokrotnie opór w miejscu zwężenia,
praktycznie nie wpływa na całkowity opór wieńcowy i spoczynkowy przepływ wieńcowy
w sercu. Dlatego, przepływ wieńcowy w zdrowym sercu, w uproszczeniu opisuje równanie:
Q = ΔP/Rmikrokrążenia [7]
III.2.3. Czynnościowy opór wieńcowy i jego regulacja
Opór czynnościowy zależy od napięcia mięśni gładkich budujących błonę środkową naczynia. Małe komórki mięśni gładkich (o średnicy około 3 μm, długości 30–130 μm), ułożone są okrężnie lub spiralnie w stosunku do światła naczynia. Komórki te mają zdolność
do generowania spontanicznego skurczu (rytmicznego lub stałego, zwłaszcza pod wpływem
rozciągnięcia), dzięki czemu, w naczyniu, w którym jest pewne ciśnienie, są nawet w spoczynku w stanie ciągłego napięcia. Komórki mięśniówki naczyniowej są połączone ścisłymi
złączami (gap-junctions) i tworzą syncytium elektryczne. Dlatego pobudzenie jednej komórki
(np. ukłucie igłą) rozprzestrzenia się na wszystkie pozostałe i powoduje skurcz całego fragmentu naczynia [8].
39
Na mięśniówkę gładką naczyń działają równocześnie czynniki kurczące i rozkurczające.
Dlatego ostateczny stan czynnego napięcia ściany naczyniowej (tonus naczyniowy) ustala się
w wyniku sumowania się tych efektów. Za część skurczową tonusu odpowiada głównie tzw.
regulacja miogenna, a za część rozkurczową regulacja metaboliczna, śródbłonkowa i nerwowo-humoralna. Wrażliwość na działanie wymienionych mechanizmów regulacyjnych zmienia się w zależności od kalibru naczynia. Na działanie czynników metabolicznych podatne są
najmniejsze tętniczki o średnicy <100 μm, to jest te, które mają bliski kontakt z miocytami
serca. W naczyniach rzędu 80–150 μm dominuje regulacja miogenna. Regulacja śródbłonkowa najsilniej wyrażona jest w tętniczkach o średnicy 120–200 μm. Tonus większych tętnic
regulowany jest tylko w niewielkim stopniu.
III.2.3.1. Regulacja miogenna
Regulacja miogenna polega na tym, że komórki mięśni gładkich naczyń kurczą się w odpowiedzi na rozciąganie. Oznacza to, że naczynia są stale w różnym stopniu przykurczone
i że tonus naczyniowy zwiększa się wraz ze wzrostem ciśnienia wewnątrznaczyniowego. Spadek ciśnienia krwi ma skutek przeciwny. Mechanizm ten znany jako autoregulacja przepływu
wieńcowego (rozdz. III.2.5.), zapewnia: (i) stałość przepływu wieńcowego pomimo nawet
znacznych wahań ciśnienia w aorcie oraz (ii) ochronę dystalnie położonych włośniczek przed
mechanicznym uszkodzeniem związanym z nadmiernym wzrostem ciśnienia krwi. Substancje, które całkowicie rozkurczają naczynia mikrokrążenie i likwidują tonus miogenny (np.
adenozyna, dipirydamol), znoszą autoregulację i zależność między ciśnieniem perfuzyjnym
i przepływem wieńcowym staje się liniowa [9].
III.2.3.2. Regulacja metaboliczna
Dopiero wzajemne oddziaływania między tonusem miogennym i regulacją metaboliczną
i śródbłonkową umożliwia precyzyjne dostosowywanie przepływu wieńcowego do aktualnego zapotrzebowania energetycznego mięśnia sercowego. Wzrostowi zapotrzebowania energetycznego serca towarzyszy spadek ciśnienia parcjalnego tlenu (pO2) w miokardium i wzrost
ciśnienia parcjalnego CO2 Na drodze słabo poznanego mechanizmu powoduje to rozkurcz
najbardziej obwodowych tętniczek oporowych (segment <100 μm), zmniejszenie oporu
wieńcowego i wzrost przepływu. Ocenia się, że ~40% zmienności przepływu wieńcowego
podczas wysiłku fizycznego tłumaczą zmiany prężności O2 i CO2 w miokardium [10,11].
Innym czynnikiem biorącym udział w regulacji metabolicznej przepływu wieńcowego są kanały potasowe ATP-zależne (K-ATP) w komórkach mięśni gładkich tętnic wieńcowych (rozdz.
IV.3). Ich otwarcie powoduje wypływ z komórki dodatnich jonów K+, hyperpolaryzację komórki
i jej rozkurcz. Te kanały są tonicznie otwarte w spoczynku, czego dowodzi fakt, że ich bloker (glibenklamid), podany dowieńcowo redukuje spoczynkowy przepływ wieńcowy o 10–20% i ogranicza wzrost przepływu wywołany szybkim drażnieniem serca czy niedokrwieniem. Glibenklamid nie upośledza natomiast wzrostu przepływu wieńcowego podczas wysiłku fizycznego. Oznacza to, że kanały K-ATP albo nie biorą udziału w regulacji metabolicznej, albo w przypadku ich
blokady jakieś inne czynniki przejmują ich działanie regulacyjne w czasie wysiłku fizycznym.
Adenozyna, która jest produktem rozkładu ATP (rozdz. IV.3), podana do wieńcowo, silnie rozkurcza tętniczki mikrokrążenia (o średnicy <200 μm). Nie ma jednak dowodów by
40
brała udział w regulacji przepływu wieńcowego w normalnym sercu w spoczynku czy wysiłku. Niewątpliwy jest natomiast jej udział w rozkurczu naczyń prowokowanym niedokrwieniem czy niedotlenieniem.
Innym postulowanym regulatorem może być ATP uwalniane z kardiomiocytów proporcjonalnie do obciążenia wysiłkiem. ATP prowadzi do rozkurczu naczyń wieńcowych poprzez
działanie na receptory purynergiczne PY2 na komórkach śródbłonka, co skutkuje zwiększoną
śródbłonkową produkcją NO. Istnieją przekonywujące argumenty przeciwko udziałowi prostanoidów i endoteliny w metabolicznej regulacji przepływu wieńcowego. Rolę NO i receptory β-adrenergiczne w tej regulacji omawiam poniżej.
Sumując, regulacja metaboliczna przepływu wieńcowego u człowieka nie jest do końca
poznana, ale wszystko wskazuje na to, że podlega wieloczynnikowej regulacji (tzw. redundancja), w ramach której defekt jednego z mechanizmów regulacyjnych może być skutecznie
kompensowany przez pozostałe (np. sprawna regulacja wysiłkowa przepływu wieńcowego
u osobników z dysfunkcją śródbłonkową i małą dostępnością biologiczna NO).
III.2.3.3. Regulacja śródbłonkowa
Zwiększony na drodze metabolicznej przepływ uruchamia równocześnie regulację śródbłonkową przepływu. Zapewnia ona rozkurcz tętniczek położonych proksymalnie do segmentu
naczyń wrażliwych na bodźce metaboliczne i w ten sposób częściowo przejmuje na siebie ciężar
regulacji przepływu. Umożliwia to dalszą reakcję mikrokrążenia na bodziec metaboliczny [12].
Regulacja śródbłonkowa wiąże się z aktywnością wydzielniczą komórek śródbłonka.
Lepka krew, płynąc przez naczynie ociera się o komórki śródbłonka. Siła z tym związana,
tzw. siła ścinająca (shear stress) powoduje deformację integryn (białka z rodziny cząsteczek
adhezyjnych) na błonie komórkowej komórek śródbłonka, co jest bodźcem do zwiększonego
wytwarzania przez śródbłonek licznych substancji, w tym tlenku azotu (NO), prostacykliny,
tkankowego aktywatora plazminogenu, endoteliny i innych [13].
NO jest substancją o silnym działaniu rozkurczającym naczynia, a endotelina silnie kurczy naczynia. NO hamuje wydzielanie endoteliny, dlatego w zdrowym naczyniu przeważa
rozkurczające działanie NO.
Wytwarzanie NO jest minimalne w naczyniach z małym przepływem krwi i rośnie wraz
ze wzrostem przepływu i siły ścinającej. Siła ścinająca (SS) jest funkcją przepływu (Q), lepkości krwi (η) i promienia naczynia (r) w trzeciej potędze
SS = 4Qη/Πr3) [8]
Dlatego taka sama zmiana przepływu powoduje znacznie większą zmianę siły ścinającej
i uwalniania NO w małych niż w dużych naczyniach. Wzrost przepływu pod wpływem bodźca metabolicznego sprawia, że w narządzie zwiększają się: siła ścinająca, produkcja NO przez
śródbłonek i rozkurcz naczyń. Efekty te są szczególnie duże w obwodowych segmentach drzewa naczyniowego (segment o średnicy 80–150 μm). Skutkuje to dalszym zmniejszeniem oporu naczyniowego i zwiększeniem przepływu krwi, co jest efektem częściowo wzmacniającym,
a częściowo „odciążającym” mechanizm metabolicznej regulacji przepływu, a także zabezpieczającym śródbłonek przed mechanicznym uszkodzeniem przez zwiększoną siłę ścinającą.
W prawidłowym krążeniu naczynia stale produkują pewne ilości NO i znajdują się pod
„tonicznym” naczyniorozszerzającym działaniem NO. Natomiast dysfunkcja śródbłonka,
41
której towarzyszy upośledzenie śródbłonkowego wytwarzania NO zależnego od przepływu,
powoduje trwały przykurcz naczyń (zwłaszcza segmentu 80–150 μm) (wzrost oporu tego
segmentu), co pośrednio ogranicza zakres metabolicznej regulacji przepływu. Dodatkowym
powodem tego przykurczu jest zwiększona produkcja endoteliny i wywołany przez nią skurcz
naczyń oraz stres oksydacyjny [14].
Ważnym regulatorem siły ścinającej i śródbłonkowej produkcji NO, jest wielkość przepływu krwi. Wzrostowi przepływu towarzyszy wzrost siły ścinającej i produkcji NO. Naczynie się rozkurcza (rośnie promień naczynia, równanie [7]) i siła ścinająca ulega normalizacji.
W ten sposób śródbłonkowa regulacja przepływu utrzymuje siłę ścinającą na stałym poziomie, co chroni śródbłonek przed jego mechanicznym uszkodzeniem.
Liczne substancje czynne (aminy katecholowe, acetylocholina, serotonina, ADP, histamina,
endotelina) działają rozkurczająco lub kurcząco na naczynia w zależności od stanu czynnościowego śródbłonka. Działają one równocześnie na śródbłonek, gdzie stymulują produkcję NO,
i bezpośrednio na mięśnie gładkie, gdzie pobudzają skurcz. W naczyniu ze zdrowym śródbłonkiem ich działanie wynikające z uwalniania NO przeważa i następuje rozkurcz naczynia. Natomiast w naczyniach z uszkodzonym śródbłonkiem powodują one skurcz naczynia.
III.2.3.4. Regulacja nerwowa i humoralna
Włókna zazwojowe współczulne i przywspółczulne kończą się w błonie zewnętrznej
(przydance) naczyń wieńcowych, uwalniając odpowiednio noradrenalinę i acetylocholinę. Substancje te, a także substancje lokalnie uwalniane przez płytki krwi (serotonina, ADP
i trombokan), komórki tuczne (histamina) i komórki śródbłonka (endotelina, bradykinina)
działają na ścianę naczynia według tego samego schematu. Z jednej strony, działając bezpośrednio na mięśniówkę naczyń powodują jej skurcz. Z drugiej strony, działając na komórki
śródbłonka, pobudzają je do produkcji NO, co ma działanie naczynio-rozkurczające. Efekt
końcowy tego współzawodnictwa mechanizmów zależy głównie od stanu funkcjonalnego
śródbłonka. W zdrowym naczyniu zazwyczaj przeważa rozkurcz związany z uwalnianiem
NO przez te substancje (acetylocholina, bradykinina, histamina) lub dochodzi do niewielkiego przykurczu (noradrenalina, endotelina, tromboksan). Natomiast w obecności dysfunkcji
śródbłonka wszystkie te substancje wywołują silny skurcz naczynia [15].
Acetylocholina podana dowieńcowo wywołuje w zdrowym krążeniu rozkurcz naczyń
i wzrost całkowitego przepływu wieńcowego. Zakończenia nerwów przywspółczulnych znajdują się w przydance naczyń krwionośnych i acetylocholina wydzielana z nich dociera najpierw do
mięśni gładkich naczyń (na które działa kurcząco), a dopiero w drugiej kolejności do śródbłonka, który pobudza do uwalniania NO. W doświadczeniach, w których porównywano wpływ pobudzania nerwu błędnego i dowieńcowej acetylocholiny na przepływ wieńcowy wykazano, że
dowieńcowe podanie acetylocholiny ma silniejsze działanie na przepływ wieńcowy. Panuje opinia, że w fizjologii wpływ układu przywspółczulnego na krążenie wieńcowe jest marginalny.
Głównym neuroprzekaźnikiem układu współczulnego wpływającym na serce jest noradrenalina uwalniania z zakończeń współczulnych, podczas gdy katecholaminy pochodzące
z rdzenia nadnerczy wywierają jedynie minimalny wpływ na serce (odpowiadają za ~2% całkowitej aktywności współczulnej). Katecholaminy oddziałują na krążenie wieńcowe, bezpośrednio i pośrednio, za pomocą receptorów:
42
1. α-adrenergicznych znajdujących się na komórkach mięśni gładkich (ich pobudzenie kurczy naczynie) i na komórkach śródbłonka (ich pobudzenie prowadzi do wzrostu uwalniania NO i rozkurczu);
2. β2-adreenrgicznych znajdujących się na komórkach mięśni gładkich naczyń wieńcowych
i na komórkach śródbłonka (pobudzenie ich prowadzi do rozkurczu mięśni gładkich);
3. β1-adrenergicznych znajdujących się w na kardiomiocytach i komórkach węzła zatokowego: ich
pobudzenie, pośrednio poprzez zwiększenie częstotliwości rytmu serca i kurczliwości, zwiększa
zapotrzebowanie energetyczne serca i w tym mechanizmie zwiększa przepływ wieńcowy.
Noradrenalina rozkurcza izolowane tętniczki wieńcowe (o średnicy <100 μm) nawet przy
braku blokady receptorów α-adrenergicznych, natomiast w większych tętniczkach i tętnicach
dominuje skurcz zależny od receptorów α-adrenergicznych. Zatem ogólny efekt działania
noradrenaliny na krążenie wieńcowe to spadek oporu wieńcowego (który rezyduje głównie
w tętniczkach mikrokrążenia) i wzrost przepływu wieńcowego.
Spoczynkowa aktywność układu współczulnego w sercu jest niewielka, wobec czego
jego udział w regulacji spoczynkowego oporu i przepływu wieńcowego jest również mały.
Wskazuje na to brak zaburzeń przepływu wieńcowego w sercu przeszczepionym i wobec tego
odnerwionym. Potwierdzają to także badania u zdrowych ludzi, u których propranolol (nieselektywny bloker β-receptorów) powodował spadek przepływu wieńcowego jedynie w proporcji do spadku częstotliwości rytmu, ciśnienia krwi, kurczliwości miokardium i ostatecznie
konsumpcji O2.
Blokada receptorów β-adrenergicznych w wysiłku fizycznym ma trojakie konsekwencje:
(i) opóźnia wysiłkowy wzrost przepływu wieńcowego; (ii) sprawia, że wzrost ten jest mniejszy niż by to wynikało ze wzrostu częstotliwości rytmu serca, ciśnienia krwi i kurczliwości)
i w efekcie (iii) powoduje, że ekstrakcja O2 się zwiększa. W sumie wydaje się, że układ współczulny bierze udział w regulacji przepływu wieńcowego dopiero w czasie wysiłku fizycznego
(głównie za pośrednictwem receptorów β2), wspomagając i „wysubtelniając” regulację metaboliczną. Szacunkowe dane sugerują, że noradrenalina odpowiada za około 25% wzrostu
przepływu w trakcie wysiłku.
W spoczynku noradrenalina, działając na receptory α-adrenergiczne, słabo kurczy naczynia wieńcowe, a blokery receptorów α powodują około 10% wzrostu przepływu wieńcowego. Kurczący wpływ receptorów α jest nasilony w obecności dysfunkcji śródbłonka. Rola
kurczącego efektu stymulacji receptorów α nie jest wyjaśniona. Popularna hipoteza głosi, że
niewielki przykurcz naczyń w wysiłku, większy w warstwie podnasiedziowej (gdzie gęstość
receptorów α jest największa) zapobiega „podkradaniu” krwi przez warstwę podnasierdziową
warstwie podwsierdziowej. Dodatkowo zwężając naczynia zapobiega zbędnym oscylacjom
przepływu krwi w naczyniach wieńcowych [16,17].
Angiotensyna II, działając poprzez receptory AT1 zlokalizowane na mięśniach gładkich
tętnic wieńcowych, kurczy w warunkach in vitro zarówno tętnice nasierdziowe, jak i tętniczki mikrokrążenia. Przeciwny efekt wywiera działając poprzez receptory AT2 zlokalizowane
na komórkach śródbłonka, których pobudzenie prowadzi do zwiększonego uwalniania NO
i rozkurczu tętniczek. U ludzi nie wykazano istotnego wpływu zablokowania receptorów AT1
na przepływ wieńcowy ani w spoczynku, ani w czasie wysiłku, co sugeruje, że angiotensyna II
nie bierze udziału w regulacji perfuzji wieńcowej in vivo.
43
Przepływ wieńcowy (ml/min.)
III.2.4. Opór kompresyjny
Naczynia wieńcowe są „zatopione” w kurczącym się miokardium. Dlatego aktywność skurczowa serca może ograniczać przepływ wieńcowy w dwóch mechanizmach: (i) mięsień kurcząc się zaciska zatopione w nim naczynia oraz (ii) w fazie skurczu izowolumetrycznego, kiedy
gwałtownie rośnie ciśnienie komorowe, krew napiera na ściany komór, powodując wzrost tzw.
ciśnienia zewnątrznaczyniowego, które jest proporcjonalne do naprężenia w ścianie komór,
i które także uciska naczynia wieńcowe. Konsekwencją obu tych oddziaływań jest wzrost oporu
naczyniowego, a ściślej jego komponenty zewnątrznaczyniowej, w czasie systole i zjawisko fazowości przepływu wieńcowego (rys. 3.3.). Fazowość przepływu występuje głównie w ścianie lewej komory, gdzie przepływ jest największy w diastole a następnie gwałtownie maleje w systole.
Ucisk zewnątrznaczyniowy rośnie w tych stanach, którym towarzyszy wzrost naprężenia
w ścianie komór (miara obciążenia serca). Rośnie także wraz ze wzrostem rytmu serca. Zgodnie z prawem Laplace’a, naprężenie jest proporcjonalne do ciśnienia komorowego i odwrotnie proporcjonalne do promienia krzywizny komory (rozmiarów komory). Dlatego, zgodnie
z przestrzennym rozkładem naprężenia i oporu kompresyjnego w sercu, fazowość przepływu
jest niewielka w ścianie prawej komory. Natomiast w lewej jest duża w okolicy podwsierdziowej i maleje w kierunku nasierdzia.
Systole
Diastole
Rys. 3.3. Fazowość przepływu wieńcowego w ścianie lewej komory serca. Przepływ odbywa się głównie
w fazie rozkurczu i jest znacznie zredukowany w fazie skurczu komory.
Wiadomo także, że opór kompresyjny rośnie w niewydolnych i powiększonych sercach
(bo naprężenie w ich ścianie jest zwiększone), co ogranicza rezerwę wieńcową w tych sercach.
Ucisk zewnątrznaczyniowy ogranicza przepływ wieńcowy głównie w czasie systole. Dlatego,
w pewnych warunkach, przyspieszenie rytmu może ograniczać przepływ wieńcowy. Wynika
to z faktu, że wraz ze wzrostem rytmu serca rośnie frakcja czasu, jaką serce spędza w fazie
systole kosztem skrócenia diastole (ryc. 3.4.).
Całkowity opór wieńcowy jest sumą oporu czynnego i kompresyjnego. W sercach z normalnymi tętnicami wieńcowymi efekty oporu kompresyjnego na przepływ wieńcowy są kompensowane częściowym rozkurczem mięśniówki gładkiej naczyń (co skutkuje redukcją czynnego oporu naczyniowego) i przepływ wieńcowy pozostaje niezmieniony. Innymi słowy, by
utrzymać stały przepływ, serce „poświęca” część rezerwy wazodylatacyjnej na skompensowanie
oporu kompresyjnego. Dlatego ubytek tej rezerwy jest większy w lewej niż w prawej komorze
44
Ryc. 3.4. Zależność między częstością akcji serca i długością fazy rozkurczu lewej komory u psów, którym podawano wzrastające dawki iwabradyny w czasie spoczynku i podczas wysiłku fizycznego. Ponieważ zależność ta jest nieliniowa, niewielkie zwolnienie rytmu (np. od 100/min do 80/min, o 25%)
powoduje nieproporcjonalnie większe wydłużenie diastole (z 370 ms do 520 ms, o 40%). Zmodyfikowane wg [18]
i większy we wsierdziu niż nasierdziu. Sytuacja zmienia się radykalnie w obszarach serca bez
rezerwy wazodylatacyjnej (np. zaopatrywanych przez stenotyczną tętnicę). W takich obszarach
perfuzja wieńcowa maleje proporcjonalnie do rytmu serca lub innego czynnika zwiększającego
komponent pozanaczyniowy oporu wieńcowego, gdyż nie może on być kompensowany czynnym rozkurczem naczyń. Ilustrują to następujące obserwacje eksperymentalne:
W sercu z maksymalnie rozkurczonymi naczyniami (np. pod wpływem adenozyny)
celem eliminacji czynnościowej komponenty oporu, przepływ wieńcowy jest liniowo zależny od ciśnienia perfuzyjnego i oporu kompresyjnego. Jeżeli takie serce zostaje zatrzymane
w rozkurczu (celem eliminacji komponenty kompresyjnej oporu), przepływ w jego warstwie
podwsierdziowej jest ~30% większy niż w warstwie podnasierdziowej (bo gęstość włośniczek
w warstwie podwsierdziowej jest większa). Natomiast, kiedy jest stymulowane z wzrastającą
częstością, dochodzi do spadku przepływu w subendokardium, co jest odpowiedzią na wzrost
ucisku zewnątrznaczyniowego i opóru kompresyjnego.
Natomiast w normalnym krążeniu wieńcowym udział ucisku zewnątrznaczyniowego w regulacji przepływu jest niewidoczny. Dla przykładu, w sercu psa pobudzanym coraz szybszym rytmem stwierdzono, że przepływ wieńcowy rośnie we wszystkich warstwach miokardium mimo
że częstszej stymulacji towarzyszy wzrost oporu kompresyjnego. Chodzi o to, że częstszej stymulacji towarzyszy równoczesny wzrost konsumpcji O2. Spowodowany tym spadek czynnościowej
komponenty oporu jest większy niż wzrost jego komponenty kompresyjnej. Gdy doprowadzimy
do maksymalnego rozkurczu naczyń wieńcowych (w warunkach eksperymentalnych podając
wazodylatator, np. adenozynę; podobna sytuacja ma miejsce w miażdżycowo zmienionym krążeniu wieńcowym, w obszarze za krytyczną stenozą), opór czynnościowy staje się minimalny
i niezmienny. Wzrost częstotliwości rytmu w warstwie subepikardialnej nie zmienia przepływu
wieńcowego: oczywiście prowadzi do wzrostu zapotrzebowania tlenowego myocardium, ale opór
czynnościowy już wyjściowo był tu minimalny. Natomiast ucisk zewnątrznaczyniowy w tej war45
stwie jest nieistotny. Tymczasem w subendocardium wzrost częstotliwości rytmu i związany z tym
wzrost oporu kompresyjnego prowadzi do proporcjonalnego spadku przepływu.
Sumując, opór kompresyjny rośnie wraz z naprężeniem w komorze i rytmem serca. W zdrowym krążeniu wieńcowym jego wpływ na przepływ wieńcowy jest niewidoczny, gdyż jest kompensowany równoczesnym spadkiem oporu czynnościowego. Naprężenie i rytm serca stają się
natomiast głównymi determinantami przepływu wieńcowego w stanach, którym towarzyszy
znaczna redukcja komponenty czynnościowej oporu (np. obszar za stenozą wieńcową).
III.2.5. Autoregulacja przepływu wieńcowego i rezerwa wieńcowa
W zdrowym sercu, wbrew równaniu nr [1], zwiększanie ciśnienia perfuzyjnego w krążeniu wieńcowym powoduje proporcjonalny wzrost przepływu wieńcowego jedynie w zakresie
bardzo niskich i bardzo wysokich ciśnień perfuzyjnych. Natomiast w pośrednim przedziale
ciśnień przepływ wieńcowy jest praktycznie stały (ryc. 3.5.). Proces, w którym mimo zmian
ciśnienia perfuzyjnego, przepływ wieńcowy pozostaje niezmieniony, nazywamy autoregulacją przepływu wieńcowego. U podłoża tego procesu leżą zmiany napięcia miogennego mięśni
gładkich naczyń i współuczestniczące zmiany regulacji metabolicznej.
Ryc. 3.5. Wpływ ciśnienia perfuzyjnego na przepływ wieńcowy w lewej komorze serca. (A) – przepływ
w normalnym sercu; (M) – przepływ maksymalny po podaniu wazodylatatora. R75 i R100 – rezerwa wieńcowa przy ciśnieniu perfuzyjnym odpowiednio 75 i 100 mmHg.
Regulacja miogenna nie działa poniżej dolnego punktu granicznego autoregulacji i tętniczki oporowe mikrokrążenia są maksymalnie rozkruczone. Takie całkowicie rozkurczone naczynia mają dwie ważne właściwości (i) reagują na zmiany ciśnienia krwi wzrostem przepływu
zgodnie z równaniem [1] oraz (ii) główną determinantą ich przepływu jest opór kompresyjny.
W zakresie ciśnień, w którym działa autoregulacja przepływu, napięcie miogenne i komponenta czynnościowa oporu rosną proporcjonalnie do wzrostu ciśnienia perfuzyjnego i, wobec tego, przepływ pozostaje niezmieniony. Znaczenie czynnościowe autoregulacji jest dwojakie: (i) pozwala uniezależnić dostawę O2 od aktualnych wahań ciśnienia tętniczego krwi
oraz (ii) chroni włośniczki przed mechanicznym uszkodzeniem spowodowanym nadmiernym wzrostem ciśnienia krwi.
46
Powyżej górnego punktu granicznego autoregulacji, mięśniówka naczyń jest maksymalnie skurczona i wobec tego naczynie ponownie zachowuje się zgodnie z równaniem [1].
Poziom, na którym utrzymywany jest stały przepływ w mechanizmie autoregulacji (ryc.
3.5., krzywa A) jest wypadkową regulacji miogennej i metabolicznej przepływu wieńcowego.
W tym drugim wypadku chodzi o wielkość zapotrzebowania energetycznego mięśnia sercowego oraz stężenie hemoglobiny we krwi (determinującego zawartość O2 w danej objętości
krwi). Gdy zapotrzebowanie energetyczne mięśnia sercowego rośnie lub, gdy spada stężenie
hemoglobiny, włącza się metaboliczna regulacja oporu naczyniowego i plateau krzywej autoregulacji ustala się na poziomie większego przepływu.
Przepływ maksymalny (ryc. 3.5., krzywa M) jest w pełnym zakresie ciśnień perfuzyjnych zależny od ciśnienia a w warstwie podwsierdziowej dodatkowo zależy od częstotliwości
rytmu serca, która ogranicza maksymalny przepływ wieńcowy poprzez zwiększony ucisk zewnątrznaczyniowy (vide III.2.4.).
Gdy przy stałym obciążeniu serca ciśnienie w aorcie spada nagle np. z 100 mmHg do
75 mmHg, dochodzi do krótkotrwałego spadku przepływu wieńcowego zgodnie z równaniem [1]. Uruchamia to dwa przeciwdziałające temu spadkowi mechanizmy; (i) w odpowiedzi na spadek ciśnienia perfuzyjnego, spada miogenne napięcie mięśniówki gładkiej naczyń
i opór naczyniowy maleje oraz (ii) spadek przepływu i związane z tym niedokrwienie aktywują rozkurcz tętniczek wrażliwych metabolicznie, co w ciągu kilku sekund skutkuje powrotem
przepływu wieńcowy do poziomu wyjściowego.
Autoregulacja występuje w naczyniach wieńcowych z zachowanym czynnym napięciem
mięśniówki naczyniowej. Z chwilą, gdy napięcie to zostanie wyeliminowane (np. pod wpływem wazodylatatora) przepływ wieńcowy rośnie, a zależność między przepływem a ciśnieniem staje się liniowa (ryc. 3.5., krzywa M). Różnicę między przepływem spoczynkowym
i maksymalnym możliwym nazywamy rezerwą wieńcową i wyrażamy w procentach przepływu wyjściowego. U zdrowych osobników rezerwa wieńcowa wynosi 200–500% i nigdy
nie stanowi czynnika ograniczającego wysiłek fizyczny (nawet przy maksymalnych wysiłkach
naczynia wieńcowe można bardziej rozkurczyć).
Cztery czynniki decydują o wielkości rezerwy wieńcowej w sercu. Są to:
1. Ciśnienie perfuzyjne u wlotu do mikrokrążenia; jak ilustruje to ryc. 3.5., wzrost ciśnienia perfuzyjnego w zakresie autoregulacji, mimo że nie wpływa na spoczynkowy przepływ
wieńcowy, systematycznie zwiększa wielkość rezerwy wieńcowej;
2. Sumaryczna średnica naczyń oporowych mikrokrążenia w danym obszarze; im jest ona
mniejsza, tym nachylenie krzywej zależności przepływ/ciśnienie perfuzyjne w obecności
wazodilatatora (miara komponenty anatomicznej oporu naczyniowego) jest bardziej strome tym większe są przyrosty rezerwy wieńcowej spowodowanej wzrostem ciśnienia perfuzyjnego;
3. Częstotliwość rytmu serca; każde przyspieszenie rytmu skutkuje wzrostem oporu kompresyjnego. W normalnych naczyniach nie towarzyszy temu ograniczenie spoczynkowego
przepływu wieńcowego a jedynie ograniczenie rezerwy wieńcowej (rozdz. III.2.4.);
4. Przepływ spoczynkowy; im bardziej w spoczynku rozkurczone są tętniczki oporowe mikrokrążenia i im większy jest w sercu przepływ spoczynkowy, tym serce dysponuje mniejszą rezerwą wieńcową. Widać to na przykładzie niedokrwistości, w przebiegu której spa47
dek stężenia hemoglobiny wymusza wzrost spoczynkowego przepływu wieńcowego, co
zmniejsza rezerwę wieńcową.
Warstwa podwsierdziowa ściany lewej komory ma mniejszą rezerwę wieńcową i węższy zakres autoregulacji niż warstwa podnasierdziowa (ryc. 3.6.). Jest to pochodna dwóch zjawisk: po
pierwsze spoczynkowa konsumpcja tlenu i przepływ spoczynkowy w subendocardium są o ~15%
wyższe, niż w subepicardium. Po drugie przy fizjologicznym rytmie serca (60–90/min) przepływ
maksymalny w subendocardium jest o ok. 20% niższy. Te czynniki sprawiają, że w zdrowym sercu rezerwa wieńcowa jest o około 30% niższa w warstwie podwsierdziowej niż w podnasierdziowej (ryc. 3.6.). Co ciekawe, gęstość naczyń w warstwie podwsierdziowej jest znacznie wyższa,
co sprawia, że w sercu nie kurczącym się rezerwa wieńcowa w warstwie podwsierdziowej jest
o około 50% wyższa niż w warstwie podnasierdziowej.
Rys. 3.6. Wpływ ciśnienia perfuzyjnego na przepływ wieńcowy w warstwie podwsierdziowej i podnasierdziowej lewej komory serca. W warstwie podnasierdziowej przepływ spoczynkowy jest wyższy,
przepływ maksymalny i rezerwa wieńcowa niższe i dolna granica autoregulacji położona wyżej, niż
w warstwie podnasierdziowej.
Dodatkowo zakres autoregulacji w warstwie podwsierdziowej sięga do niższych ciśnień niż
w warstwie podnasierdziowej. W efekcie dolna granica autoregulacji w warstwie podnasierdziowej wynosi około 30–40 mmHg i rośnie do około 55–65 mmHg w okolicy wsierdzia (ryc. 3.5.).
Różnice te nabierają szczególnego znaczenia w sytuacji, kiedy dochodzi do spadku ciśnienia
perfuzyjnego (np. w segmencie za stenozą). Dla przykładu, w zakresie ciśnień perfuzyjnych
30–60 mmHg przepływ w warstwie podwsierdziowej maleje prawie do zera, natomiast w warstwie podnasierdziowej pozostaje ciągle niezmieniony. Przepływu podnasierdziowego jest wyraźnie zmniejszony dopiero przy spadku ciśnienia perfuzyjnego poniżej 30 mmHg.
We wszystkich sytuacjach, w których dochodzi do zwiększenia spoczynkowej perfuzji
wieńcowej (przerost, niedokrwistość), dolna granica autoregulacji przesuwa się w kierunku
wyższych ciśnień. Zwiększa to dodatkowo zagrożenie warstwy podwsierdziowej w obszarach
zaopatrywanych przez stenotyczne naczynia, w których ciśnienia perfuzyjne są znacznie obniżone [19].
48
III.3. Przepływ wieńcowy w prawej komorze
Powyższe rozważania dotyczą przede wszystkim krążenia wieńcowego lewej komory.
Przynajmniej dwa czynniki składają się na specyfikę krążenia wieńcowego w prawej komorze. W komorze tej:
1. ekstrakcja O2 w spoczynku nie jest maksymalna (jak w lewej komorze), wobec czego jej
konsumpcja O2 może wzrastać także w mechanizmie zwiększonej ekstrakcji O2 (mniejsza
zależność od regulacji przepływu);
2. ucisk wewnątrznaczyniowy i wobec tego opór kompresyjny, są minimalne.
W efekcie, regulacja przepływu wieńcowego w prawej komorze jest bardzo podobna do
regulacji przepływu w mięśniach szkieletowych.
III.4. Regulacja przepływu wieńcowego w wysiłku
Wysiłkowi fizycznemu towarzyszy równoczesny wzrost kompresyjnej składowej oporu
naczyniowego (bo rosną częstotliwości rytmu serca i naprężenia w ścianie komory) oraz
znacznie większy spadek oporu czynnościowego związany z aktywacją regulacji metabolicznej, a następnie regulacji śródbłonkowej przepływu. W efekcie całkowity opór naczyniowy maleje, a przepływ wieńcowy proporcjonalnie wzrasta (ryc. 3.1.B). Sekwencja wydarzeń jest następująca. Początkowo, w odpowiedzi na spadek pO2 w miokardium (bardziej
obciążone kardiomiocyty zużywają więcej O2), rozkurczają się tętniczki oporowe sąsiadujące z kardiomiocytami (segment <100 μm), spada całkowity opór wieńcowy i następuje
natychmiastowy proporcjonalny wzrost przepływu wieńcowego. Uruchamia to zależną od
przepływu regulację śródbłonkową przepływu (rozdz. III.2.3.3.) skutkującą zwiększonym
uwalnianiem NO i rozkurczem głównie tętniczek położonych proksymalnie do segmentu
wrażliwego na bodźce metaboliczne (segment 120–200 μm), co ma ważne konsekwencje
czynnościowe:
1. następuje dalszy spadek oporu mikrokrążenia, zwiększenie przepływu i ponowny wzrost
tonusu miogennego (ponieważ wobec spadku oporu mikrokrążenia rośnie lokalne ciśnienie perfuzyjne), co jest efektem częściowo wzmacniającym, a częściowo „odciążającym”
mechanizm metabolicznej regulacji przepływu (ryc. 3.7.);
2. siła ścinająca, zwiększona w wyniku metabolicznego wzrostu przepływu, normalizuje się,
co chroni śródbłonek przed mechanicznym uszkodzeniem.
W prawidłowym krążeniu naczynia znajdują się pod stałym naczyniorozszerzającym
działaniem NO. Natomiast dysfunkcja śródbłonkowa, której towarzyszy upośledzenie śródbłonkowej produkcji NO zależnej od przepływu, powoduje trwały przykurcz segmentu naczyniowego 120–200 μm (i ewentualnie większych naczyń), wzrost ich oporu i kompensacyjny rozkurcz bardziej dystalnej części mikrokrążenia oraz zmniejszenie rezerwy wieńcowej
całego serca. Składają się na to następujące czynniki:
1. brak komponentu rozkurczowego zależnego od NO;
2. zwiększona wrażliwość naczyń na działanie kurczące amin katecholowych, acetylocholiny,
serotoniny, ADP, histaminy i endoteliny – prawidłowo substancje te stymulują uwalnianie
NO przez śródbłonek (działanie rozkurczające), natomiast w naczyniach z uszkodzonym
śródbłonkiem przeważa ich działanie kurczące;
49
Ryc. 3.7. Gra naczyniowa indukowana wzrostem zapotrzebowania miokardium na O2. Faza I – następuje rozkurcz tętniczek wrażliwych metabolicznie (TWM) (czerwone pole), co skutkuje spadkiem oporu
naczyniowego R (np. o 50%) i wzrostem przepływu. Faza II – Wzrost przepływu skutkuje wzrostem
siły ścinającej i produkcji NO oraz rozkurczem w segmentach proksymalnych do TWM i ponownym
przykurczem TWM. Dzięki „wędrówce” wazodylatacji od TWM do większych naczyń, te ostatnie przejmują ciężar regulacji przepływu, a TWM odzyskują zdolność do reakcji na bodziec metaboliczny. Wędrowanie wazodylatacji jest osłabione lub nie zachodzi w obecności dysfunkcji śródbłonkowej. Ciężar
regulacji przepływu spoczywa wtedy na TWM, które wyczerpują swój potencjał rozkurczowy już przy
małych obciążeniach. Upośledza to zdolność dostosowywania się perfuzji wieńcowej do obciążenia.
Mechanizm ten jest prawdopodobnie przyczyną kardiologicznego zespołu X.
3. wzrost śródbłonkowej produkcji endoteliny o silnym działaniu kurczącym naczynia (NO
hamuje produkcję endoteliny);
4. zwiększona naczyniowa produkcja wolnych rodników tlenowych, co jeszcze bardziej
zmniejsza dostępność biologiczną i/lub produkcję NO.
III.5. Perfuzja miokardium w obecności różnych klas zwężeń tętnic
wieńcowych
Najczęstszą przyczyną zwężeń są blaszki miażdżycowe, które występują wyłącznie w tętnicach nasierdziowych, które to tętnice mają pomijalny opór naczyniowy. Zwężenie powoduje
wzrost oporu tętnicy nasierdziowej, spadek ciśnienia za zwężeniem i spadek efektywnego ciśnienia perfuzyjnego w obszarze za stenozą. Skutkuje to w tym obszarze: (i) spadkiem miogennego
tonusu mięśniówki naczyń oporowych; (ii) rozkurczem tych naczyń i spadkiem ich oporu; (iii)
niezmiennym przepływem spoczynkowym mimo obecności stenozy oraz (iv) zmniejszeniem
rezerwy wazodiltacyjnej w obszarze za stenozą. Ze względu na konsekwencje dla regulacji przepływu wieńcowego, wyróżnia się trzy klasy zwężeń/stenoz tętnic nasierdziowych (ryc. 3.8.).
Klasa I (zwężenia nieistotne) – zmniejszenie średnicy tętnicy lub pola jej przekroju nie przekracza odpowiednio 50% i 75%. Zmiany tego typu nie zwiększają jeszcze oporu tętnicy nasierdziowej i wobec tego pozostają bez wpływu na hemodynamikę krążenia wieńcowego, zarówno
w spoczynku jak i w czasie wysiłku (chyba, że stają się źródłem ostrego incydentu wieńcowego);
Klasa II (zwężenia istotne) – ograniczenie średnicy lub pola przekroju tętnicy o odpowiednio 50–80% i 75–90%; zwężenia te zwiększają opór tętnicy nasierdziowej, ale w stopniu,
50
Ryc. 3.8. Wpływ wielkości zwężenia tętnicy nasierdziowej (Klasy I, II i III) na jej opór (RS) oraz opór
mikrokrążenia (RM) i spoczynkowy przepływ (linia czerwona) w jej dorzeczu. Szczegóły w tekście.
który może zostać skompensowany przez zmniejszenie oporu czynnościowego mikrokrążenia. Zwężenia istotne powodują proporcjonalny do ich oporu:
1. spadek ciśnienia za zwężeniem (do 100–60 mm Hg);
2. odpowiednie zmniejszenie ciśnienia perfuzyjnego w mikrokrążeniu;
3. zmniejszenie tonusu miogennego w mikrokrążeniu i spadek jego oporu czynnościowego.
W efekcie tych zmian całkowity opór wieńcowy, i tym samym spoczynkowy przepływ
wieńcowy, pozostają niezmienione, ale odbywa się to kosztem wyczerpywania się rezerwy
wieńcowej w obszarze zaopatrywanym przez zwężoną tętnicę (ryc. 3.1.C) Niekorzystne konsekwencje ujawniają się dopiero w czasie wysiłków fizycznych (p. niżej i ryc. 3.1.D).
Klasa III (zwężenia krytyczne) – ograniczenie średnicy lub pola przekroju tętnicy o odpowiednio >80% i >90%. Opór takich zwężeń jest tak duży, że ciśnienie za zwężeniem maleje
do wartości <50–60 mmHg, stanowiących dolną granicę autoregulacji krążenia wieńcowego
(ryc. 3.8.). W tych warunkach rezerwa rozkurczowa mikrokrążenia jest już całkowicie wyczerpana i proporcjonalnie do zmniejszenia ciśnienia perfuzyjnego (wielkości zwężenia) następuje ograniczenie przepływu wieńcowego nawet w spoczynku.
Klasyfikacja stenoz powstała w oparciu o wyniki eksperymentów na zwierzętach, w których stenozy wywoływano poprzez stopniowe zaciskanie podwiązki na zdrowej tętnicy. U ludzi rozwój stenozy jest procesem długotrwałym i towarzyszy mu rozwój krążenia obocznego
i dysfunkcji śródbłonkowej. Dlatego zakresy zwężeń typowe dla poszczególnych klas, mogą
być różne u ludzi i zwierząt.
Przyczyną wysiłkowego niedokrwienia mięśnia sercowego w stabilnej chorobie wieńcowej
są najczęściej zwężenia klasy II. W ich obecności spoczynkowy przepływ wieńcowy jest normalny, natomiast rezerwa wieńcowa jest, w znacznej mierze, wyczerpana, bardziej w warstwie podwsierdziowej niż podnasierdziowej lewej komory (ryc. 3.1.C) (V.2.). W czasie wysiłku (podobnie jak w normalnym sercu), rośnie opór kompresyjny i dochodzi do aktywacji metabolicznej
regulacji przepływu. Jednakże rozkurcz metaboliczny następuje głównie w warstwie podnasierdziowej, a w warstwie podwsierdziowej jest bardzo mały, lub nieobecny. W rezultacie całkowity
51
opór naczyniowy maleje w warstwie podnasierdziowej i pozostaje niezmieniony lub wzrasta
w warstwie podwsierdziowej, co przekierowuje krew płynącą przez zwężoną tętnicę nasierdziową do warstwy podnasierdziowej kosztem ograniczenia przepływu w warstwie podwsierdziowej (efekt podkradania). Ostatecznie w czasie wysiłku w obszarze serca zaopatrywanym przez
istotnie zwężoną tętnicę wieńcową od pewnego poziomu nasilenia wysiłku:
1. przepływ w warstwie podnasierdziowej rośnie;
2. przepływ w warstwie podwsierdziowej albo nie rośnie proporcjonalnie do wysiłku, albo,
w obecności bardziej zaawansowanych stenoz, nawet maleje (ryc. 3.1.D) i obie te sytuacje
skutkują niedokrwieniem (vide V.2);
3. interwencje zmniejszające opór kompresyjny, zwłaszcza zmniejszające częstotliwość rytmu serca, zmniejszają niekorzystną redystrybucję przepływu z warstwy powsierdziowej do podnasierdziowej, poprawiając tolerancję wysiłku u chorych na stabilną chorobę wieńcową (VI.2).
III.6. Krążenie wieńcowe w nadciśnieniu tętniczym
W nadciśnieniu tętniczym powszechnie obserwuje się obniżenie rezerwy wieńcowej. Odpowiedzialne są za to przynajmniej cztery mechanizmy patofizjologiczne:
1. Nadciśnienie prowadzi do niekorzystnych zmian struktury tętnic wieńcowych (m.in. przerostu ich ściany);
Ryc. 3.9. Przepływ wieńcowy w trzech warstwach przedniej ściany lewej komory (EPI, warstwa podnasierdziowa; MID, warstwa środkowa; ENDO, warstwa podwsierdziowa) u zdrowych psów (lewy panel) i psów
z przerostem lewej komory (prawy panel). Czarne kółka – przepływ w spoczynku; białe kółka – przepływ w wysiłku; czarne trójkąty – przepływ w spoczynku w obecności adenozyny; białe trójkąty – wysiłek
w obecności adenozyny. W spoczynku stosunek przepływów end/epi wynosi 1,5 w kontroli i tylko 1,1
w przeroście. W wysiłku stosunek przepływów endo/epi obniżył się nieznacznie u kontrolnych zwierząt
(1,3) i znacznie w przeroście (0,8). Spoczynkowa rezerwa wieńcowa w kontroli i przeroście (stosunek przepływu w obecności adenozyny do przepływu spoczynkowego) wynosiła ~400% i była podobna we wszystkich warstwach lewej komory (stosunek endo/epi ~1,0). Natomiast w czasie wysiłku stosunek przepływów
endo/epi spadł do 0,8 w sercach kontrolnych i do 0,4 w sercach z przerostem [20]
52
2. Nadciśnieniu towarzyszy zaburzenie czynności naczyń wieńcowych pod postacią dysfunkcji śródbłonkowej;
3. U większości chorych, elementem obrazu nadciśnienia tętniczego jest przerost lewej komory,
co prowadzi do wzrostu komponentu pozanaczyniowego oporu wieńcowego i jeszcze większego ubytku rezerwy wazodylatacyjnej, szczególnie w czasie wysiłku fizycznego, w co najmniej dwóch mechanizmach: (i) w przerośniętym sercu w miarę zwiększania częstotliwości
rytmu serca dochodzi do nieproporcjonalnie większego skrócenia fazy rozkurczu, w porównaniu z sercem nieprzerośniętym; (ii) spadek podatności lewej komory sprawia, że związany
z wysiłkiem wzrost objętości końcoworozkurczowej odbywa się kosztem znacznego wzrostu
ciśnienia rozkurczowego w lewej komorze (nawet do 30 mmHg). To sprawia, że nie dość, iż
faza rozkurczu jest krótsza, to jeszcze w jej trakcie naczynia wieńcowe są uciskane z zewnątrz
(ryc. 3.9.). Z tego powodu zaburzenia krążenia wieńcowego u osób z nadciśnieniem tętniczym bardzo często występują w wysiłku, zwłaszcza w warstwie podwsierdziowej;
4. Nadciśnienie jest czynnikiem ryzyka miażdżycy i związanych z nią zaburzeń krążenia
wieńcowego.
III.7 Kardiologiczny zespół X – zaburzenia mikrokrążenia wieńcowego
Kardiologiczny zespół X (dławica piersiowa bez zmian w dużych tętnicach wieńcowych)
to postać choroby wieńcowej obecna u 10–20% pacjentów poddawanych koronarografii,
w której występują dolegliwości w klatce piersiowej, dodatnia próba obciążeniowa, natomiast
duże tętnice wieńcowe nie wykazują istotnych zwężeń.
Patofizjologia kardiologicznego zespołu X nie jest do końca poznana. Za główną przyczynę uznaje się zaburzenia mikrokrążenia wieńcowego, które mogą towarzyszyć zwężeniu dużej
tętnicy nasierdziowej (dodatkowo przyczyniając się do redukcji rezerwy wieńcowej w dorzeczu tetnicy), ale mogą też występować jako izolowana patologia (ryc. 3.10.) [21].
Ryc. 3.10. Zaburzenia mikrokrążenia wieńcowego (żółte koło) mogą towarzyszyć stenotycznej postaci choroby wieńcowej, dodatkowo przyczyniając się do obniżenia rezerwy wieńcowej w tym obszarze.
Mogą także występować jako izolowana patologia, odpowiadając za przynajmniej część przypadków
kardiologicznego zespołu X.
U około 70% osób z tak zdefiniowanym zespołem X, mimo dodatnich wyników próby
wysiłkowej nie stwierdza się biochemicznych cech niedokrwienia (pomiar stężenia kwasu
53
mlekowego, produktów peroksydacji lipidów czy saturacji hemoglobiny we krwi w zatoce
wieńcowej), co sugeruje, że źródłem dolegliwości niekoniecznie jest niedokrwienie mięśnia
sercowego [22]. Jedną z przyczyn dolegliwości w kardiologicznym zespole X mogą być zaburzenia percepcji bólu (V.4).
Znane są dwie patofizjologiczne postacie zaburzeń mikrokrążenia:
1. spowodowana dysfunkcją śródbłonka tętniczek mikrokrążenia – prowadzi do upośledzenia rozkurczu naczyń mikrokrążenia w czasie wysiłku i do wtórnego obniżenia rezerwy
wieńcowej. Podkreśla się związek tej postaci choroby z hipercholesterolemią zwłaszcza, że
jej objawy ustępują pod wpływem leczenia hipolipemizującego;
2. związana z nadmierną skłonnością naczyń mikrokrążenia do skurczu; u części pacjentów
z zaburzeniami mikrokrążenia występują zaburzenia przepływu wieńcowego pod wpływem potencjalnie kurczących czynników: hiperwentylacji, wysiłku umysłowego, ergonowiny, przy braku skurczu tętnic nasierdziowych, oraz paradoksalny spadek przepływu pod
wpływem wysiłku fizycznego [21].
W stenotycznej postaci choroby wieńcowej, zaburzenia perfuzji wieńcowej ograniczają
się najczęściej do okolicy podwsierdziowej dorzecza zwężonej tętnicy i można je zidentyfikować bezpośrednio – przy pomocy badania scyntygraficznego lub pośrednio – w postaci lokalnych zaburzeń kurczliwości w trakcie echokardiograficznego testu obciążeniowego (rozdz.
III.8.). Natomiast w przypadku patologii mikrokrążenia, zaburzenia perfuzji, jeżeli w ogóle
daje się je zidentyfikować w badaniu scyntygraficznym, mają charakter rozsiany i zwykle nie
powodują zaburzeń kurczliwości identyfikowalnych w echokardiografii [22].
III.8. Czynnościowe metody oceny istotności hemodynamicznej stenozy
Techniki obrazowania (koronarografia, TK) pozwalają na identyfikację zwężeń tętnic wieńcowych, ale często zawodzą, kiedy przychodzi do oceny czynnościowego znaczenia danego zwężenia. Bardziej precyzyjnych informacji dostarczają wtedy tzw. czynnościowe testy obciążeniowe.
Ogólna zasada polega na prowokowaniu w sercu maksymalnego rozkurczu naczyń mikrokrążenia albo pod wpływem wazodilatatora (np. dipiridamol) albo wysiłku fizycznego czy dobutaminy
(poprzez wzrost kurczliwości miokardium i przyspieszenie akcji serca powodują metaboliczny
wzrost perfuzji wieńcowej), a następnie na pomiarze różnymi technikami perfuzji wieńcowej
w różnych obszarach mięśnia lewej komory (scyntygrafia perfuzyjna), lub konsekwencji niedokrwienia (próba wysiłkowa EKG, echokardiograficzna próba obciążeniowa z dobutaminą).
III.8.1. Scyntygrafia perfuzyjna (SPECT)
W badaniu SPECT (emisyjna tomografia komputerowa pojedynczego fotonu) do pomiaru perfuzji miokardium wykorzystuje się promieniotwórcze znaczniki perfuzji takie jak tal
(201Tl) i technet (99mTc). Substancje te są w sposób czynny wychwytywane przez kardiomiocyty z krwi, proporcjonalnie do wielkości perfuzji (w pewnym zakresie, ryc. 3.11.). Wobec tego
ich regionalny wychwyt jest funkcją wielkości lokalnej perfuzji wieńcowej, ale także żywotności kardiomiocytów. Interpretacja badania opiera się na analizie lokalnych różnic promieniotwórczości w obrębie lewej komory mięśnia sercowego. Badanie nie dostarcza informacji na
temat bezwzględnej wielkości perfuzji w danym obszarze (w ml/min/g tkanki).
54
Stwierdzenie zmniejszonego wychwytu znacznika w jakimś obszarze miokardium w spoczynku (tzw. nieodwracalny ubytek perfuzji) sugeruje albo obecność (i) obszaru bez żywych
kardiomiocytów albo (ii) obszaru z zachowaną żywotnością kardiomiocytów, ale zmniejszoną spoczynkową perfuzją (np. mięsień hibernowany i/lub ogłuszony).
Znaczny ubytek wychwytu znacznika sugeruje istnienie segmentu martwiczego i scyntygrafia (99mTc sestamibi) należy od uznanych metod oceny wielkości zawału. Podanie znacznika
przed reperfuzją pozwala na określenie wielkości obszaru niedokrwionego (tzw. area at risk).
Powtórne podanie znacznika po reperfuzji pozwala na określenie obszaru martwiczego. Różnica
wielkości tych dwóch obszarów jest miarą wielkości obszaru uratowanego przez reperfuzję.
Ryc. 3.11. Liniowa zależność między przepływem wieńcowym i wychwytem znacznika promieniotwórczego przy umiarkowanych przepływach i brak zależności przy wyższych przepływach.
Może się zdarzyć, że w badaniu spoczynkowym perfuzja całego miokardium jest podobna, i że różnice perfuzji ujawniają się dopiero w czasie testu obciążeniowego (wysiłek, dobutamina lub wazodilatator). Obecność ubytków perfuzji tylko w czasie testu obciążeniowego
(tzw. odwracalne ubytki perfuzji) oznacza, że w analizowanym obszarze: (i) kardiomiocyty
są żywe; (ii) spoczynkowa perfuzja jest prawidłowa oraz (iii) perfuzja w badanym obszarze
jest mniejsza niż w pozostałym mięśniu (technika nie pozwala na ocenę czy wielkość perfuzji zmalała, wzrosła, czy nie zmieniła się w porównaniu z perfuzją spoczynkową), co razem
wziąwszy oznacza, że rezerwa wieńcowa w badanym obszarze jest zmniejszona.
III.8.2. Farmakologiczny test obciążeniowy z wazodilatatorem
Zamiast obciążać serce wysiłkiem czy dobutaminą, można stosować substancje farmakologiczne, których efekty na krążenie wieńcowe naśladują zmiany wysiłkowe. Zaletą testów
z wazodylatatorami jest to, że powodują one większe wzrosty przepływu niż wysiłek fizyczny czy dobutamina (bo tachykardia towarzysząca wysiłkowi czy dobutaminie zwiększa opór
kompresyjny i ogranicza rezerwę wieńcową).
Najczęściej stosowane są adenozyna i dypirydamol (bloker transportera zwrotnego wychwytu adenozyny powodujący wzrost tkankowego stężenia adenozyny). Adenozyna, działając przez
receptor A2 w mięśniach gładkich tętniczek oporowych mikrokrążenia wieńcowego, powoduje
ich rozkurcz. Tętniczki oporowe w obszarach za stenozą (ze zmniejszoną rezerwą wieńcową) są
częściowo rozkurczone już w spoczynku. Dlatego wazodylatator spowoduje większy rozkurcz na55
czyń mikrokrążenia w normalnym miokardium niż w obszarze za stenozą. Będzie to skutkowało
mniejszym wzrostem przepływu w obszarze za stenozą i regionalnymi różnicami w przepływie,
widzianymi jako różnice w wychwycie znaczników promieniotwórczych (odwracalne ubytki perfuzji). U 10–15% pacjentów w trakcie farmakologicznego testu obciążeniowego, niedobory przepływu w badanym obszarze są tak duże, że dochodzi do obniżenia odcinka ST lub zaburzeń kurczliwości miokardium, obserwowanych echokardiograficznie, świadczących o niedokrwieniu.
III.8.3. Regionalna rezerwa wieńcowa (fractional flow reserve, FFR)
FFR jest inwazyjną techniką oceny istotności hemodynamicznej (czynnościowej) stenozy
wieńcowej (ryc. 3.8.) bazującą na fakcie, że w obecności stenozy rośnie opór dla przepływu
krwi i dochodzi do spadku ciśnienia krwi za stenozą. FFR oblicza się ze wzoru:
Pdyst
FFR = -------- [7]
Pproks
Pdyst – ciśnienie za stenozą; Pproks – ciśnienie przed stenozą
Różnica ciśnień w poprzek stenozy (ΔP = Q x R) jest proporcjonalna do wielkości oporu
naczyniowego (R), na który składa się głównie opór stawiany przez stenozą ale częściowo także opór obwodowego mikrokrążenia. Wobec tego pomiary FFR wykonywane są w obecności
wazodilatatora, w stężeniu powodującym maksymalny rozkurcz naczyń [23]. W ten sposób
zostaje zminimalizowana komponenta oporu naczyniowego zależna od mikrokrążenia i opór
wynikający z obecności stenozy staje się głównym czynnikiem determinującym wartości gradientu ciśnień w poprzek stenozy.
W zdrowej tętnicy nasierdziowej nie ma istotnego spadku oporu na jej przebiegu (Pdyst =
Pproks) i wobec tego jej FFR przyjmuje wartość 1,0. Wraz z narastaniem zwężenia tętnicy i towarzyszącym spadkiem Pdyst wartość FFR spada poniżej jedności. Czynnościowo oznacza to,
że narastanie zwężenia tętnicy i wynikły z tego spadek ciśnienia perfuzyjnego w jej dorzeczu
(Pdyst), skutkują kompensacyjnym rozkurczem mikrokrażenia i spadkiem rezerwy wazodilatacyjnej w obszarze za stenozą (ryc. 3.8.).
Natomiast na drodze empirycznych obserwacji i porównań ustalono, że FFR < 0,75–0,80
jest „markerem” czynnościowej istotności zwężenia tętnicy wieńcowej (ryc. 3.8.). Im wartość
FFR jest mniejsza, tym większa jest istotność hemodynamiczna stenozy.
FFR ma szereg przewag nad innymi technikami oceny istotności hemodynamicznej zwężeń tętnic wieńcowych (koronarografią, IVUS, lub koronarografią TK):
1. FFR jest miarą całkowitego oporu, jaki stawia badane zwężenie (parametr fizjologiczny o bezpośrednim znaczeniu dla zaburzeń perfuzji) i tym samym uwzględnia takie zmienne jak długość zwężenia (im większa tym opór większy) i regularność jego obrysu (zwężeniom nieregularnym towarzyszy burzliwy przepływu krwi dodatkowo zwiększający opór naczyniowy);
2. FFR jest niewrażliwy na artefakty związane z dwuwymiarową oceną trójwymiarowej struktury w koronarografii;
3. FFR uwzględnia obecność ewentualnego krążenia obocznego, które może sprawiać, że
nawet duże zwężenie będzie z czynnościowego punktu widzenia nieistotne (im krążenie
oboczne lepsze tym spadek oporu na stenozie mniejszy).
56
Ograniczeniem FFR jest fakt, że jest miarą istotności zwężenia jedynie dużych tętnic nasierdziowych i nie pozwala ocenić czy i na ile do zaburzeń rezerwy wieńcowej przyczyniają się
nieprawidłowości w obrębie mikrokrążenia wieńcowego występujące w dorzeczu zwężonej
tętnicy. Dodatkowe ograniczenie wynika z faktu, że cewnik pomiarowy sam dodatkowo ogranicza światło naczynia w miejscu zwężenia, co może skutkować przeszacowaniem wielkości
zweżenia, zwłaszcza w przypadku małych naczyń z dużymi zwężeniami.
W badaniu FAME porównywano wartość pomiaru FFR (FFR ≤0,80) i koronarografii jako
podstawy kwalifikacji pacjentów z wielonaczyniową chorobą wieńcową do wszczepiania stentów
(uwalniających lek przeciwproliferacyjny) [24]. Okazało się, że w grupie kwalifikowanej do stentowania w oparciu o FFR zużyto mniej stentów i w ciągu roku obserwacji, rzadziej wystąpił w niej
pierwszorzędowy punkt końcowy (zgon, zawał niezakończony zgonem, powtórna rewaskularyzacja), co podkreśla wartość tej techniki w ocenie zaawansowania zmian miażdżycowych.
III.8.4. Echokardiografia obciążeniowa
W metodzie tej ocenia się zmianę kurczliwości miokardium podczas stopniowo rosnącego obciążenia mięśnia sercowego, a w praktyce najczęściej w obecności wzrastających dawek dobutaminy
(agonista receptorów β-adrenergicznych i słaby agonista α-receptorów). Interpretacja echokardiografii obciążeniowej opiera się na trzech zasadach fizjologicznych: (1) zarówno segmenty martwicze
jak i hibernowane i ogłuszone wykazują zaburzenia kurczliwości w spoczynku; (2) tylko segmenty
z zachowaną żywotnością (hibernowane, ogłuszone) reagują zmianą kurczliwości na dobutamine;
(3) kierunek zmiany kurczliwości (wzrost vs. spadek) zależy od wielkości rezerwy wieńcowej w tętnicy zaopatrującej badany segment i zdolności perfuzji wieńcowej do nadążania za zwiększonym
obciążeniem miokardium. W praktyce obserwowane są cztery wzory reakcji na dobutaminę:
A. podanie dobutaminy nie skutkuje zmianą kurczliwości badanego segmentu, co sugeruje
brak w nim żywotnego miokardium;
B. Wzraz ze wzrostem dawki dobutaminy rośnie kurczliwość segmentu; sugeruje to obecność
żywotnego miokardium w badanym segmencie (ogłuszenie lub hibernacja) oraz to, że segment jest zaopatrywany w tętnicę bez istotnego zwężenia (w badanym zakresie obciążeń
przepływ dostosowuje się do obciążenia);
C. Wraz ze wzrostem dawki dobutaminy kurczliwość segmentu najpierw rośnie a przy wyższych dawkach maleje (tzw. odpowiedź dwufazowa); sugeruje to żywotność miokardium
w badanym segmencie oraz to, że jest on zaopatrywany przez istotnie zwężoną tętnicę
wieńcową (z prawidłowym przepływem spoczynkowym i ograniczoną rezerwą wieńcową). W obecności małych stężeń dobutaminy przepływ wieńcowy jeszcze nadąża za wzrastającym obciążeniem, co umożliwia wzrost kurczliwości. W obecności wyższych stężeń
(większego obciążenia miokardium) rezerwa wieńcowa ulega wyczerpaniu, przepływ nie
nadąża za obciążeniem i dochodzi do niedokrwienia o typie niedokrwienia wysiłkowego.
W efekcie, kurczliwość segmentu maleje proporcjonalnie do stopnia niestosunku między
obciążeniem i perfuzją (V.5.1). Im mniejsza jest dawka dobutaminy skutkująca spadkiem
kurczliwości tym istotność hemodynamiczna stenozy jest większa;
D. Nawet mała dawka dobutamine skutkuje pogorszeniem kurczliwości badanego segmentu.
Fakt, że mięsień zareagował zmianą kurczliwości oznacza, że zachował żywotność. Fakt, że
kurczliwość zmalała już w obecności niewielkiego obciążenia sugeruje, że tętnica zaopa57
trująca segment jest zwężona w stopniu krytycznym (rezerwa wieńcowa w segmencie jest
całkowicie wyczerpana i zwiększonemu obciążeniu nie towarzyszy już wzrost przepływu).
Piśmiennictwo
1. Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiol Rev 2008; 88:1009-1086.
2. Feigl EO: Coronary physiology. Physiol Rev 1983; 63:1-110.
3. Kassab GS, Rider CA, Tang NJ, Fung YC. Morphometry of pig coronary arterial trees. American
Journal of Physiology - Heart and Circulatory Physiology 1993; 265:H350-H365.
4. Hoffman JIE, Spaan JAE: Pressure-flow relations in coronary circulation. Physiol Rev 1990; 70:331-389.
5. Epstein SE, Cannon RO,III, Talbot TL. Hemodynamic principles in the control of coronary blood
flow. Am. J. Cardiol. 1985, 56, 4E-10E.
6. Defily DV, Chilian WM. Coronary microcirculation: autoregulation and metabolic control. Basic
Res. Cardiol. 1995, 90, 112-118.
7. Muller JM, Davis MJ, Chilian WM. Integrated regulation of pressure and flow in the coronary microcirculation. Cardiovasc. Res. 1996; 32: 668-678.
8. Liu Y, Gutterman DD: Vascular control in humans: Focus on the coronary microcirculation. Basic
Res Cardiol 2009; 104:211-227.
9. Miller FJ, Dellsperger KC, Gutterman DD: Myogenic constriction of human coronary arterioles. Am
J Physiol Heart Circ Physiol 1997; 273:H257-H264.
10. Kanatsuka H, Lamping KG, Eastham CL, et al. Comparison of the effects of increased myocardial oxygen
consumption and adenosine on the coronary microvascular resistance. Circ Res 1989; 65:1296-1305.
11. Tune JD, Gorman MW, Feigl EO. Matching coronary blood flow to myocardial oxygen consumption.
J Appl Physiol 2004; 97:404-415.
12. Stepp DW, Nishikawa Y, Chilian WM: Regulation of shear stress in the canine coronary microcirculation. Circulation 1999; 100:1555–1561.
13. Kuo L, Davis MJ, Chilian WM: Endothelium-dependent, flow-induced dilation of isolated coronary
arterioles. Am J Physiol 1990; 259:H1063-H1070.
14. Chilian WM, Layne SM, Klausner EC, et al. Redistribution of coronary microvascular resistance
produced by dipyridamole. Am J Physiol 1989; 256:H383-H392.
15. Chierchia, S., L. Muiesan, A. Davies, V et al. Role of the sympathetic nervous system in the pathogenesis of chronic stable angina. Implications for the mechanism of action of beta-blockers. Circulation 1990, 82 (Suppl II):II-71-II-81.
16. Sun D, Huang A, Mital S et al. Norepinephrine Elicits {beta}2-Receptor-Mediated Dilation of Isolated Human Coronary Arterioles. Circulation 2002; 106:550-555.
17. Heusch G, Baumgart D, Camici P, et al. α-Adrenergic coronary vasoconstriction and myocardial
ischemia in humans. Circulation 2000; 101:689-699.
18. Colin P, Ghaleh B, Monnet X, Hittinger L, Berdeaux A. Effect of graded heart rate reduction with
ivabradine on myocardial oxygen consumption and diastolic time in exercising dogs. J Pharmac Exp
Therap 2004; 308:236-240.
19. Maczewski M, Beręsewicz A. Krążenie wieńcowe i jego zaburzenia w chorobie wieńcowej. w.: red. Beręsewicz
A, Dłużniewski M, Metaboliczne leczenie choroby wieńcowej. Gdańsk, Via MEdica 2006: 14-44.
20. Duncker, DJ, Ishibashi Y i Bache RJ. Effect of treadmill exercise on transmural distribution of blood
flow in hypertrophied left ventricle. Am J Physiol Heart Circ Physiol 1998; 275: H1274-H1282.
21. Lanza GA. Cardiac syndrome X: a critical overview and future perspectives. Heart 2007; 93:159-166.
22. Lanza GA, Crea F. Primary coronary microvascular dysfunction: Clinical presentation, pathophysiology, and management. Circulation 2010; 121:2317-2325.
23. Spaan JA, Piek JJ, Hoffman JI, et al. Physiological basis of clinically used coronary hemodynamic
indices. Circulation 2006; 113:446-458.
24. Tonino PAL, De Bruyne B, Pijls NHJ et al. Fractional flow reserve versus angiography for guiding
percutaneous coronary intervention. New England Journal of Medicine 2009; 360:213-24.
58
IV. Przemiany substratów energetycznych
w sercu
Andrzej BERĘSEWICZ
IV.1. Ogólny schemat metabolizmu energetycznego
Energia do produkcji wysokoenergetycznych wiązań reszt kwasu fosforowego w ATP
pochodzi z reakcji łączenia się tlenu (O2) z wodorem w mitochondrialnym łańcuchu oddechowym. Tlen jest rozpuszczony w płynach ustrojowych i jest stosunkowo łatwo dostępny.
Natomiast źródłem wodoru są tzw. substraty energetyczne serca, czyli cząsteczki niezestryfikowanych kwasów tłuszczowych (NEFA), glukozy, kwasu mlekowego i ewentualnie ketonów.
Tlenowa produkcja ATP polega na tym, że w przebiegu kolejnych reakcji enzymatycznych,
wodór jest stopniowo odłączany od substratów energetycznych i przenoszony najpierw na
kofaktory NAD i FAD a następnie z ich zredukowanych form (NADH i FADH2) na O2 (ryc.
4.1.).
Ryc. 4.1. Tlenowe przemiany substratów energetycznych jako źródło ATP dla skurczu i transportu jonów.
59
(A)
(B)
Ryc. 4.2. Metabolizm energetyczny kwasów tłuszczowych, glukozy i kwasu mlekowego w mięśniu sercowym. A. Wspólnym produktem wstępnych przemian tych substratów jest acetylo-CoA. W cyklu Krebsa
zostaje on utleniony i powstają zredukowane kofaktory NADH i FADH2. W procesie fosforylacji oksydacyjnej protony są przenoszone z tych kofaktorów na O2 czemu towarzyszy synteza ATP (rozdz. V.8.1).
W prawidłowo utlenowanym sercu, preferowanym substratem są kwasy tłuszczowe, gdyż produkty
ich przemian hamują metabolizm glukozy na poziomie: błonowego transportu glukozy, glikolizy oraz
utleniania pirogronianu (linie przerywane). MCT-1, transporter kwasu mlekowego; Glut-4, transporter
glukozy; FAT/CD36, translokaza długo-łańcuchowych kwasów tłuszczowych; HK, heksokinaza; PFK-1,
fosfofruktokinaza-1, CPT-I, transferaza karnityno-palmitynowa I; TG, wewnątrzkomórkowe triglicerydy; ANT, translokaza nukleotydów ATP i ADP. B. Transport ATP z mitochondriów do aparatu kurczliwego następuje z udziałem tarnslokazy ATP/ADP (ANT) oraz kreatyny (Cr) i fosfokreatyny (CrP).
CPK-MI i CPK-MM – mitochondrialna i miofibrylarna izoforma kinazy kreatynowej (CPK).
60
Proces tlenowej produkcji ATP składa się z następujących etapów (ryc. 4.2.) [1]:
1. NEFA, glukoza i kwas mlekowy dopływają do kardiomiocytów z krwią i przechodzą do
cytoplazmy przy udziale odpowiednich transporterów błonowych [2,3]. Glukoza i NEFA
mogą pochodzić także z ich wewnątrzkomórkowych magazynów, jakimi są glikogen i triglicerydy;
2. W komórce, NEFA, glukoza i kwas mlekowy są dzielone na dwuwęglowe fragmenty (reszty kwasu octowego), które w obecności koenzymu A (CoA) tworzą acetylo-CoA, główny
substrat cyklu Krebsa. Acetylo-CoA powstaje z glukozy w procesie glikolizy i utleniania
pirogronianu (rozdz. IV.3.), a z NEFA – w procesie β-oksydacji (rozdz. IV.4.);
3. Cykl kwasów trójkarboksykowych (cykl Krebsa); proces, w którym od acetylo-CoA odłączany jest wodór i przenoszony na kofaktory NAD i FAD. Powstają wtedy ich zredukowane formy (NADH i FADH2) oraz woda i dwutlenek węgla (stąd powiedzenie, że substraty energetyczne są spalane w cyklu Krebsa). Obok cyklu Krebsa, NADH powstaje jeszcze
w procesach β-oksydacji NEFA i spalania pirogronianu, a FADH2 w procesie β-oksydacji;
4. Fosforylacja oksydacyjna – wieloelementowy proces, w którym protony są przenoszone z NADH i FADH2 na O2 i energia wtedy wyzwalana służy do produkcji ATP (rozdz.
V.8.1);
5. Transport ATP z macierzy mitochondrialnej do przestrzeni międzybłonowej mitochondriów przy udziale translokazy ATP/ADP, a następnie transport ATP do cytoplazmy i aparatu kurczliwego kardiomiocytów z udziałem kreatyny, gdzie ATP zasila skurcz i inne ATPzależne procesy.
IV.2. Transport ATP z mitochondriów do aparatu kurczliwego
ATP jest transportowany z macierzy mitochondrialnej do przestrzeni między wewnętrzną i zewnętrzną błoną mitochondrialną przez białko translokazę ATP/ADP. Obecna tam mitochondrialna izoforma kinazy kreatynowej (CPKmito) przenosi wysokoenergetyczny fosforan
z ATP na kreatynę i powstaje fosfokreatyna i ADP (ryc. 4.2.B). Natomiast ADP wraca do
macierzy mitochondrialnej, gdzie jest ponownie fosforylowany do ATP. Fosfokreatyna, której
cząsteczka jest wielokrotnie mniejsza od ATP, dyfunduje do aparatu kurczliwego kardiomiocyta. Miofibrylarna izoforma CPK (CPKMM) katalizuje tam reakcję przeniesienia wysokoenergetycznego fosforanu z fosfokreatyny na ADP i odtworzenie ATP, który jest już bezpośrednio
dostępny dla aparatu kurczliwego. Natomiast wolna kreatyna wraca do mitochondriów po
kolejną resztę fosforanową.
Kreatyna produkowana przez wątrobę i nerki jest transportowana do miocytów, wbrew
gradientowi stężeń, przez błonowy transporter kreatyny. Około 75% kreatyny obecnej w sercu występuje w postaci fosfokreatyny, a 25% jako wolna kreatyna. Niewielkie ilości kreatyny
są stale tracone przez kardiomiocyty na drodze biernej dyfuzji.
System kreatyna-fosfokreatyna zapewnia szybki transport ATP do aparatu kurczliwego.
Dodatkowo fosfokreatyna jest zapasowym magazynem energii przeciwdziałającym niepożądanym spadkom komórkowego stężenia ATP. W sytuacjach, kiedy mitochondrialna produkcja ATP nie nadąża za jego rozkładem (nagły wzrost obciążenia, niedokrwienie), cytoplazmatyczne stężenie ATP pozostaje przez pewien czas niezmienione, kosztem przyspieszonego
61
rozkładu fosfokreatyny. Dlatego w sercach z niedoborem kreatyny i upośledzonym kreatynowym transportem ATP, interwencje inotropowe skutkują mniejszym wzrostem kurczliwości
mięśnia sercowego, a niedokrwienie jej szybszym spadkiem [4,5]. W niewydolności serca,
komórkowe poziomy ATP i fosfokreatyny są obniżone odpowiednio o 20% i 60%, co sprawia, że stosunek stężeń fosfokreatyna/ATP również maleje. Sugeruje to, że ważną przyczyną
zaburzeń kurczliwości w tym zespole jest deficyt energii i/lub niezdolność kardiomiocytów
do elastycznego dostosowywania produkcji i/lub transportu ATP do aktualnego obciążenia.
Stosunek fosfokreatyna/ATP jest lepszym wykładniem ryzyka zgonu w niewydolności serca
niż klasyfikacja NYHA lub frakcja wyrzucania [4].
IV.3. Przemiany glukozy i kwasu mlekowego
W warunkach tlenowych, glukoza i kwas mlekowy dostają się do komórek przy udziale
odpowiednich transporterów błonowych (ryc. 4.2.A). O szybkości tego transportu decydują
przezbłonowy gradient stężeń substancji oraz gęstość transporterów na powierzchni błony
komórkowej. W sercu występują dwie izoformy transportera glukozy, GLUT 1 i GLUT 4, ale
jedynie gęstość GLUT 4 podlega regulacji. GLUT 4 są obecne częściowo w błonie komórkowej
a częściowo magazynowane w pęcherzykach podbłonowych w cytoplazmie. Wysiłek fizyczny
i niedokrwienie (poprzez aktywację kinazy AMP, rozdz. IV.7) oraz insulina (poprzez aktywację kinazy Akt) (rozdz. IV.6), przyspieszają dokomórkowy transport glukozy gdyż powodują
przemieszczanie się GLUT 4 z pęcherzyków na powierzchnię błony komórkowej [2,3].
Wolna glukoza ulega w cytoplazmie natychmiastowej fosforylacji (przy udziale heksokinazy) i powstaje glukozo-6-fosforan, dzięki czemu glukoza nie może już opuścić komórki.
Glukozo-6-fosforan staje się substratem do syntezy glikogenu, albo podlega procesowi glikolizy.
Glikolizą nazywamy sekwencję kilku reakcji enzymatycznych, w których z jednej cząsteczki glukozo-6 fosforanu powstają 2 cząsteczki kwasu pirogronowego (w warunkach beztlenowych 2 cząsteczki kwasu mlekowego) oraz 2 cząsteczki ATP i produkcja ta nie wymaga
obecności O2 (ryc. 4.3.)
Ryc. 4.3. Całkowite przemiany energetyczne glukozy dostarczają 38 cząsteczek ATP. Z tego jedynie 2
cząsteczki (5%) powstają w wyniku glikolizy, co nie wystarcza do podtrzymywania skurczu. Komórkowa lokalizacja enzymów glikolitycznych sugeruje, że ATP z glikolizy zasila głównie transport Ca2+
w siateczce śródplazmatycznej i rozkurcz miocytów oraz czynność elektryczną serca [1]
62
Pirogronian przechodzi do macierzy mitochondrialnej (przy udziale transportera błonowego w wewnętrznej błonie mitochondrialnej) gdzie ulega dalszemu spalaniu. W normalnym
sercu, w spoczynku, kwas pirogronowy spalany przez serce w połowie pochodzi z glikolizy
a w połowie z kwasu mlekowego przekształcanego w kwas pirogronowy przez dehydrogenazę kwasu mlekowego. Podczas wysiłku, kiedy rośnie produkcja kwasu mlekowego w mięśniach szkieletowych, może on stać się głównym substratem energetycznym serca. W mitochondriach kwas pirogronowy jest dekarboksylowany przez dehydrogenazę pirogronianową
(PDH) i powstały w ten sposób acetylo-CoA jest utleniany w cyklu Krebsa do CO2, czemu
towarzyszy produkcja dalszych 36 cząsteczek ATP (ryc. 4.3.).
W normalnym sercu procesy glikolizy i utleniania glukozy są z sobą ściśle sprzężone, co
oznacza, że całość kwasu pirogronowego powstającego w procesie glikolizy jest na bieżąco
usuwana z cytoplazmy i spalana w mitochondriach. W niedokrwieniu a także w reperfuzji,
PDH, z różnych powodów, nie nadąża za glikolityczną produkcją kwasu pirogronowego. Stan
ten, znany jako rozprzęganie glikolizy i utleniania glukozy, skutkuje komórkową akumulacją
kwasu pirogronowego, który następnie przekształcając się w kwas mlekowy, zakwasza komórki i ich otoczenie, z licznymi tego niekorzystnymi konsekwencjami (rozdz. V.7).
W obrębie szlaków energetycznych glukozy regulacji podlegają: (i) transport błonowy
glukozy (poprzez regulację gęstości GLUT 4 na powierzchni błony komórkowej); (ii) szybkość glikolizy (poprzez zmianę aktywności głównie heksokinazy i fosfofruktokinazy-1); (iii)
utlenianie pirogronianu (poprzez regulację aktywności dehydrogenazy pirogronianowej)
oraz (iv) synteza i rozkład glikogenu (poprzez zmianę aktywności odpowiednich enzymów).
Głównymi czynnikami środowiska modyfikującymi przemiany glukozy (i równocześnie przemiany NEFA) w sercu są: agoniści receptorów β adrenergicznych, poziomy we krwi NEFA
i insuliny (rozdz. IV.6) oraz wysiłek fizyczny (rozdz. IV.7), a także niedokrwienie i reperfuzja
(rozdz. V).
IV.4. Przemiany niezestryfikowanych kwasów tłuszczowych (NEFA)
Szybkość wychwytywania NEFA przez serce zależy od ich stężenia w krwi (rozdz. IV.5)
oraz gęstości transporterów NEFA na błonie komórkowej. Podobnie jak glukoza, także NEFA
są transportowane do komórek przy pomocy przynajmniej dwóch białkowych transporterów
błonowych FATP (fatty acid transport protein) oraz FAT/CD36 (fatty acid translocase/CD36)
i dwóch kooperujących z nimi białkami wiążącymi kwasy tłuszczowe (FABP). Stosunkowo najwięcej wiadomo o fizjologii FAT/CD36. Jest on odpowiedzialny za transport błonowy głównie
długołańcuchowych kwasów tłuszczowych. Jest częściowo obecny na powierzchni błony komórkowej a częściowo w pęcherzykach podbłonowych i jego gęstość na błonie podlega regulacji. Podobnie jak w przypadku GLUT 4 (rozdz. IV.3), wysiłek fizyczny i niedokrwienie, poprzez
aktywację kinazy AMP (rozdz. IV.7) a insulina, poprzez aktywację kinazy Akt (rozdz. IV.6),
przyspieszają dokomórkowy transport NEFA gdyż stymulują przemieszczanie się FAT/CD36
z pęcherzyków do błony komórkowej [3]. FAT/CD36 jest dodatkowo wielofunkcyjnym receptorem wymiatającym klasy B biorącym udział w rozwoju miażdżycy (rozdz. VII.1.2).
W komórce, NEFA są przekształcane, przy udziale syntetazy acylo-CoA, w estry długołańcuchowych kwasów tłuszczowych i koenzymu A (acylo-CoA, proces znany jako aktywacja
63
kwasów tłuszczowych). Około 10–30% tak powstałego acyl-CoA służy do syntezy różnych
wewnątrzkomórkowych substancji lipidowych (triacylglicerole, diacylglicerole, ceramidy)
a pozostała część (70–90%) jest transportowana do mitochondriów i spalana (ryc. 4.2.A)
[1,6].
W stanach, którym towarzyszy wzrost poziomu NEFA we krwi i ich transport do komórek rośnie, rośnie także komórkowa akumulacja triacylgliceroli, diacylgliceroli i ceramidów,
co staje się źródłem ich tzw. lipotoksyczności skutkującej, między innymi, insulinoopornością
tkanek, dysfunkcją mięśnia sercowego, czy niewydolnością serca [6,7] (rozdz. X). Triacylglicerole (triglicerydy) gromadzone w kardiomiocytach służą jako endogenne źródło kwasów
tłuszczowych i stale podlegają procesom syntezy i rozkładu, w podobny sposób jak triacylglicerole w komórkach tłuszczowych (rozdz. IV.6). Diacylglicerole są ważnym elementem sygnalizacji wewnątrzkomórkowej gdyż są naturalnym aktywatorem kinazy białkowej C. Również ceramidy mogą pełnić rolę sygnalizacyjną, między innymi aktywując apoptozę.
Transport acylo-CoA do mitochondriów odbywa się przy udziale trzech karnityno-zależnych enzymów (ryc. 4.4.). W pierwszym etapie, transferaza karnityno-palmitynowa (CPT-I)
przenosi NEFA z acylo-CoA na karnitynę i w przestrzeni pomiędzy zewnętrzną i wewnętrzną
błoną mitochondrialną powstaje acylo-karnityna. Następnie enzym translokaza acylo-karnityny (CAT) transportuje acylo-karnitynę przez wewnętrzną błonę do macierzy mitochondrialnej na wymianę z wolną karnityną. Tam, transferaza karnityno-palmitynowa II (CPT-II)
przenosi NEFA z karnityny ponownie na koenzym A i dopiero powstający w macierzy mitochondrialnej acylo-CoA może ulegać β-oksydacji.
Ryc. 4.4. Schemat metabolizmu niezestryfikowanych kwasów tłuszczowych w mięśniu sercowym. Schemat pokazuje mechanizm transportu acylo-CoA z cytoplazmy do macierzy mitochondrialnej (prawa
strona) oraz regulacji tego transportu przez poziom malonylo-CoA w cytoplazmie. CPT-I i CPT-II,
transferazy karnityno-palmitynowe I i II; CAT, translokaza karnitynoacylowa; ACC, karboksylaza acetylo-CoA; MCD, dekarboksylaza malonylo-CoA; LPL, lipaza lipoproteinowa; AMPK, kinaza AMP.
64
Transport NEFA do mitochondriów jest regulowany na poziomie CPT-I, w mechanizmie
ujemnego sprzężenia zwrotnego, w którym przemiany NEFA same się ograniczają (ryc. 4.4.).
Polega to na tym, że wraz ze wzrostem przemian NEFA, rośnie w komórce poziom acetyloCoA. Substancja ta jest przekształcana w malonylo-CoA (przez karboksylazę acetylo-CoA,
ACC), który z kolei jest silnym inhibitorem CPT I i transportu NEFA do mitochondriów.
Dekarboksylaza malonylo-CoA (MCD) szybko przywraca poziom malonylo-CoA do stanu
wyjściowego i przywraca aktywność CPT I i transport mitochondrialny NEFA. Aktywność
ACC ulega zahamowaniu (a spalanie NEFA jest aktywowane) w wyniku jej fosforylacji przez
kinazę AMP, między innymi pod wpływem wysiłku fizycznego oraz w wyniku niedokrwienia/reperfuzji (rozdz. IV.7.) [6]. Istnieją zachęcające próby stosowania farmakologicznych
inhibitorów CPT-I i MCD w kardioprotekcji niedokrwionego i reperfundowanego mięśnia
sercowego [6,8,9] (rozdz. VI.2.4).
IV.5. Preferencje substratowe i ich wpływ na efektywność mechaniczną
serca
To, który z substratów staje się głównym źródłem ATP zależy od warunków, takich jak
wysiłek fizyczny, poziom hormonów, przepływ wieńcowy, poziom substratów we krwi, czy
stan sytości/głodu.
NEFA hamują szlaki metaboliczne glukozy (ryc. 4.2.A) [1,6]. Zjawisko to ma znaczenie praktyczne gdyż w stanach, w których przemiany NEFA rosną kosztem spalania glukozy
i kwasu mlekowego, maleje równocześnie sprawność mechaniczna serca. Są tego przynajmniej dwie przyczyny. Jak pokazuje Tab. 4.1., spalanie kwasu palmitynowego (C16H32O2) dostarcza więcej ATP niż spalanie glukozy (C6H12O6) czy kwasu mlekowego (C3H6O3). Niemniej
jednak, koszt tlenowy produkcji cząsteczki ATP z kwasu palmitynowego i innych NEFA jest
o ~13% większy w porównaniu z produkcją ATP z glukozy i kwasu mlekowego. Dodatkowo,
NEFA rozprzęgają fosforylację oksydacyjną, co oznacza, że mitochondria „trwonią” O2 służący do produkcji ATP, co potencjalnie może skutkować mniejszą produkcją ATP. W sercach
prawidłowo utlenowanych i z prawidłową rezerwą wieńcową, taki tlenowo „nieoszczędny”
proces produkcji ATP z NEFA jest jednak zjawiskiem bezpiecznym i korzystnym, gdyż dostarcza duże ilości ATP. Natomiast w sercach z ograniczoną rezerwą wieńcową i tym samym
zagrożonych niedokrwieniem, intensywny metabolizm NEFA może pogarszać bilans energetyczny miokardium. Farmakologiczne przekierowanie metabolizmu serca z metabolizmu
NEFA na metabolizm glukozy może ten bilans poprawiać.
W klasycznym badaniu na psach, Mjøs wykazał, że wzrost wychwytywania NEFA przez
serce (zwiększano stężenie NEFA w surowicy do 3 mM) powodował wzrost konsumpcji O2
o 26%, czemu nie towarzyszył wzrost kurczliwości serca [10]. Podobny spadek efektywności
mechanicznej serca, spowodowany wzrostem stężenia NEFA w surowicy, potwierdziły późniejsze badania [11]. Równocześnie wykazano, że zahamowanie β-oksydacji NEFA w sercu
zmniejsza MVO2, i że towarzyszy temu wzrost utleniania glukozy i efektywności mięśnia sercowego [11]. Powyższe obserwacje stanowią teoretyczną podstawę leczenia stabilnej choroby
wieńcowej przy pomocy substancji, które przestawiają metabolizm energetyczny serca ze spalania NEFA na spalanie glukozy [1,9,12] (rozdz. VI.2.4).
65
Tabela 4.1. Koszt tlenowy produkcji ATP z cząsteczki glukozy, kwasu mlekowego i kwasu
palmitynowego (wg [11])
Szlak metaboliczny
Powstające
cząsteczki ATP
Zużywane
cząsteczki O2
Stosunek
ATP/O2
Glikoliza
Utlenianie glukozy (C6H12O6)
Utlenianie kw. mlekowego (C3H6O3).
Utlenianie kw. palmitynowego (C16H32O2)
2
36
18
129
0
6
3
25
6,0
6,0
5,16
W warunkach fizjologicznych, średniodobowe utlenianie NEFA, glukozy i kwasu mlekowego jest źródłem, odpowiednio, 60–70%, 20% i 10% ATP produkowanego w sercu. Ta preferencja substratowa wynika z faktu, że produkty metabolizmu NEFA blokują (ryc. 4.2.A):
1. transport błonowy glukozy;
2. szybkość glikolizy – poprzez hamowanie kluczowego enzymu glikolitycznego jakim jest
fosfofruktokinaza-1; czynnikiem blokującym jest kwas octowy, którego komórkowe poziomy rosną w wyniku przemian NEFA;
3. spalanie pirogronianu w mitochondriach na poziomie dehydrogenazy pirogronianowej
(PDH); w sytuacjach kiedy β-oksydacja kwasów tłuszczowych jest nasilona, w matrix mitochondrialnym rosną stężenia acetylo-CoA i NADH oraz stosunek NADH/NAD+ i acetylo
CoA/CoA i zmiany te skutkują zahamowaniem PDH i, w konsekwencji, zahamowaniem spalania glukozy i kwasu mlekowego. Farmakologiczne hamowanie utleniania kwasów tłuszczowych skutkuje wzrostem aktywności PDH i utleniania glukozy i kwasu mlekowego. Efekt ten
stanowi podstawę działania kardioprotekcyjnego interwencji metabolicznych stosowanych
w leczeniu choroby niedokrwiennej serca [1,9,12] (rozdz. VI.2.4).
Ciała ketonowe stają się preferowanym substratem w takich stanach jak źle kontrolowana
cukrzyca, głodzenie, czy przewlekła niewydolność serca, w których, w związku ze wzrostem
poziomu NEFA we krwi, rośnie wątrobowa produkcja ciał ketonowych. Podobnie jak NEFA,
ciała ketonowe hamują utlenianie glukozy na poziomie PDH. Dodatkowo hamują także utlenianie NEFA, ale mechanizm tego działania jest słabo poznany [1].
IV.6. Poziomy gukozy i NEFA w surowicy regulatorami metabolizmu
serca; rola insuliny
O wyborze substratu energetycznego przez serce decyduje proporcja stężeń NEFA i glukozy we krwi, warunkowana głównie przez insulinę.
Insulina jest ważnym regulatorem metabolizmu energetycznego serca (i innych narządów) gdyż: (i) poprzez działanie na tkankę tłuszczową, zmniejsza poziom NEFA we krwi
i ekspozycję na nie komórek sercowych (ryc. 4.5.) oraz (ii) działając poprzez aktywację wewnątrzkomórkowych kinaz białkowych, w tym kinazy PKB/Akt, bezpośrednio modyfikuje
aktywność komórkowych szlaków metabolicznych glukozy i NEFA (ryc. 4.6.).
Poziom NEFA we krwi jest pochodną wielkości uwalniania NEFA z komórek tłuszczowych. Komórki te stale wychwytują NEFA z krwi i magazynują je w postaci triglicerydów
(przy udziale transferazy acylo-glicerolo-3 fosforanu) i równocześnie stale rozkładają trigli66
cerydy (przy udziale hormonozależnej lipazy). Netto uwalnianie NEFA z komórek tłuszczowych jest wynikiem sumowania się tych dwóch, przeciwstawnych procesów. Insulina aktywuje transferazę acylo-glicerolo-3 fosforanu i hamuje hormonozależną lipazę. Lipaza ta jest
natomiast aktywowana przez katecholaminy (via β-receptory) i ten efekt jest odpowiedzialny
za wzrost poziomu NEFA we krwi w stanach, którym towarzyszy aktywacja współczulna (np.
niedokrwienie mięśnia sercowego) [6].
(A)
(B)
Ryc. 4.5. Regulacja stężenia NEFA we krwi i metabolizmu energetycznego serca w stanie sytości i głodzenia. (A) Po posiłku, kiedy poziomy insuliny są wysokie a NEFA niskie, głównym źródłem sercowego
ATP jest spalanie glukozy, a NEFA są raczej magazynowane w kardiomiocytach jako triglicerydy, niż
spalane. (B) Wraz z upływem czasu od posiłku (ale także w czasie głodzenia, w cukrzycy, niewydolności
serca i w czasie aktywacji współczulnej), kiedy poziomy insuliny są niskie a katecholamin wysokie i kiedy poziom NEFA jest wysoki, głównym źródłem sercowego ATP jest utlenianie NEFA, a glukoza jest
raczej magazynowana w postaci glikogenu niż spalana. Grubość linii wskazuje na intensywność danego
procesu metabolicznego.
Tuż po posiłku, kiedy stężenie glukozy we krwi wzrasta, rośnie także wydzielanie insuliny
(ryc. 4.5.A). Hiperglikemia i hiperinsulinemia, poprzez hamowanie hormonozależnej lipazy
i promowanie syntezy triglicerydów, sprawiają, że magazynowanie NEFA w tkance tłuszczowej wzrasta a ich poziom we krwi maleje. Maleje wobec tego podaż NEFA do komórek sercowych i ich preferowanym substratem do produkcji ATP staje się glukoza. W miarę upływu
czasu od posiłku, poziomy insuliny maleją a katecholamin rosną, i rosną również poziomy
NEFA (nawet czterokrotnie od 0,2 do 0,8 mM) (ryc. 4.5.B). W efekcie NEFA stają się preferowanym substratem. Podobnie dzieje się w takich stanach jak źle kontrolowana cukrzyca,
niewydolność serca, głodzenie czy aktywacja współczulna, w których stężenia NEFA we krwi
mogą rosnąć powyżej 1 mM, [1].
Insulina wywiera działanie wewnątrzkomórkowe poprzez aktywację różnych kinaz. Działanie pro-przerostowe insuliny jest związane głównie z aktywacją szlaku MAP-kinaz. Natomiast jej efekty metaboliczne są wtórne do aktywacji szlaku kinazy PI3K, aktywującej kinazy
PKB/Akt oraz fosforylacji szeregu metabolicznie ważnych białek przez tę kinazę [2]. W konsekwencji, pod wpływem insuliny dochodzi do przekierowania metabolizmu energetycznego kardiomiocytów z preferencyjnego spalania NEFA na preferencyjne spalanie glukozy
67
(ryc. 4.6.). Składają się na to następujące efekty: (i) zwiększone przechodzenie GLUT 4
z pęcherzyków podbłonowych do błony komórkowej i przyspieszony dokomórkowy transport glukozy; (ii) pośrednia aktywacja syntazy glikogenu i jego zwiększone magazynowanie
w kardiomiocytach; (iii) pośrednia aktywacja fosfofruktokinazy-1 i w konsekwencji całego
procesu glikolizy i, wtórnie do zwiększonej podaży pirogronianu do mitochondriów, aktywacja mitochondrialnego procesu spalania glukozy; (iv) zwiększona ekspresja transportera
NEFA (FAT/CD36) na powierzchni błony komórkowej i przyspieszony dokomórkowy transport NEFA (ale poziom NEFA we krwi jest obniżony w obecności insuliny, co ogranicza
ich transport dokomórkowy) oraz (v) zwiększone magazynowanie NEFA wchodzących do
komórek w postaci triglicerydów, co ogranicza spalanie NEFA i hamowanie przez nie metabolizmu glukozy.
Ryc. 4.6. Wpływ hiperglikemii i insuliny na metabolizm mięśnia sercowego. Hiperglikemia prowokuje
zwiększone wydzielanie insuliny. W jej obecności zasadniczym źródłem ATP staje się utlenianie glukozy. Insulina zwiększa, bowiem dokomórkowy transport glukozy, i aktywność glikolizy. Równocześnie
zwiększa dokomórkowy transport NEFA, ale są one magazynowane w postaci triacylgliceroli (szczegóły
w tekście). Przerywane strzałki wskazują wewnątrzkomórkowe procesy/enzymy regulowane przez insulinę. Grubość linii wskazuje na intensywność danego procesu.
Kwasica towarzysząca niedokrwieniu blokuje komórkowe efekty insuliny. Są one jednak
ciągle obecne w okresie reperfuzji. I tego okresu prawdopodobnie dotyczy potencjalne, ale
ciągle niepewne klinicznie, kardioprotekcyjne działanie mieszaniny glukoza-insulina-potas
68
(GIK). Możliwe są dwa mechanizmy takiego działania GIK [2]. Po pierwsze, poprzez przekierowanie metabolizmu mięśnia sercowego na bardziej „tlenowo-oszczędny” metabolizm
glukozy (rozdz. IV.5), GIK i sama insulina mogą poprawiać bilans energetyczny reperfundowanego miokardium. Po drugie, aktywacja kinazy PKB/Akt przez insulinę ma różne pozametaboliczne i potencjalnie kardioprotekcyjne efekty (kardioprotekcyjne działanie hartowania
niedokrwieniem jest wtórne do aktywacji kinazy PKB/Akt, rozdz. VI.3). Najważniejsze z nich
to: (i) hamowanie różnych elementów procesu apoptozy; (ii) fosforylacja i aktywacja śródbłonkowej syntazy tlenku azotu (eNOS) (zwiększona produkcja NO ma działanie protekcyjne
na naczynia i kardiomiocyty wtórne do NO-zależnej produkcji cyklicznego GMP i aktywacją
kinaz białkowej PKG); (iii) stymulacja reperfuzyjnej syntezy białek kurczliwych kardiomiocytów. Podobny profil kardioprotekcyjnego działania jak insulina ma także insulino-podobny
czynnik wzrostowy (IGF-1).
IV.7. Równowaga energetyczna serca podczas wysiłku
– rola kinazy AMP
Serce nie ma zapasów energii. Dlatego warunkiem prawidłowego działania pompy sercowej jest to by produkcja ATP w sercu na bieżąco dostosowywała się do jego zużycia. W zdrowym sercu mechanizm ten działa sprawnie i komórkowe poziomy ATP pozostają na stałym
poziomie, nawet w czasie znacznych wysiłków fizycznych. Dzieje się tak, dlatego że czynnikami „sprzęgającymi” produkcję z rozkładem ATP w sercu są: (i) komórkowe poziomy
Ca2+ (wyzwalają skurcz i determinują wielkość zużycia ATP przez aparat kurczliwy) oraz (ii)
komórkowe poziomy ADP i AMP. W tym ostatnim wypadku chodzi o to, że kiedy ATP jest
defosforylowany do ADP zgodnie z reakcją: ATP p ADP + Pi , enzym kinaza adenylowa częściowo regeneruje ATP zgodnie z reakcją: 2ADP ' ATP + AMP i wobec tego poziomy ADP
i AMP są markerami statusu energetycznego komórek.
Aktywatorami spalania mitochondrialnego i fosforylacji oksydacyjnej są dwa czynniki: (i) cytoplazmatyczny poziom ADP i Pi – substancje te są transportowane do macierzy
mitochondrialnej przy udziale translokazy ATP/ADP (ryc. 4.2.A), co dostarcza do macierzy
zwiększonych ilości substratu do produkcji ATP przez syntazę ATP a równocześnie zwiększa
transport ATP z macierzy do cytoplazmy (rozdz. IV.2); (ii) poziom Ca2+ w cytoplazmie – Ca2+
jest regulatorem rozkładu ATP i równocześnie, po wejściu do mitochondriów, aktywuje mitochondrialne dehydrogenazy (dehydrogenazę pirogronianową i dwie dehydrogenazy będące
elementem cyklu Krebsa – dehydrogenazę izocytrynianową i dehydrogenazę 2-oksoglutaronową) zwiększające ostatecznie syntezę ATP.
„Sensorem” statusu energetycznego komórek (nie tylko sercowych) i „strażnikiem” ich
równowagi energetycznej jest dodatkowo cytoplazmatyczna kinaza białkowa aktywowana
przez AMP (AMPK) [13,14]. W istocie, aktywność AMPK rośnie, kiedy rośnie komórkowy stosunek stężeń AMP/ATP, co czyni ten enzym szczególnie czułym sensorem obrotu komórkowego ATP. Dodatkowo, AMPK jest fosforylowana (i w ten sposób aktywowana) przez
dwie kinazy, z których kinaza LKB1 działa tylko w obecności AMP, a kinaza CaMKK (kinaza
kinazy zależnej od Ca-kalmoduliny) jest aktywowana przez Ca2+. Naturalnymi inhibitorami
AMPK są ATP i glikogen (ryc. 4.8.A).
69
Ryc. 4.7. Mechanizm homeostazy energetycznej serca w warunkach zwiększonego obciążenia. Lewa
strona: Wraz z obciążeniem rośnie mitochondrialna produkcja ATP gdyż: (i) zwiększony dokomórkowy
napływ Ca2+ skutkuje jego akumulacją w mitochondriach i aktywacją enzymów produkujących ATP; (ii)
zwiększony rozkład ATP skutkuje wzrostem cytoplazmatycznego stężenia ADP i Pi i ich podaży do mitochondriów, gdzie stanowią substrat dla zwiększonej syntezy ATP; Prawa strona: Równocześnie AMP
stymuluje kinazę zależna od AMP (AMPK) i wtórnie – transport dokomórkowy i przemiany NEFA
i glukozy i ostatecznie produkcję ATP
Ryc. 4.8. Regulacja aktywności AMPK (A) i procesy regulowane przez ten enzym (B). Różne czynniki, o których wiadomo, że aktywują AMPK (Ca2+, adiponektyna, wolne rodniki tlenowe, metformina)
działają na AMPK pośrednio, poprzez zmianę aktywności aktywujących ją kinaz LKB1 i/lub CaMKK.
Strzałki i linie przerywane oznaczają odpowiednio aktywację i hamowanie procesu.
Do aktywacji AMPK dochodzi w stanach fizjologicznych i patologicznych, którym towarzyszy wzrost komórkowego poziomu AMP i/lub Ca2+ (i ewentualnie spadek komórkowych
zasobów glikogenu). Skurcz mięśnia prowokuje umiarkowaną, a niedokrwienie z reperfuzją
70
i zwiekszenie obsiążenie skurczowe znaczną aktywację AMPK i jest to proces służący utrzymaniu równowagi energetycznej komórek. Aktywacja AMPK skutkuje, bowiem szybkim
wzrostem komórkowych przemian glukozy i NEFA (i zwiększoną produkcją ATP) a także
zahamowaniem energochłonnej syntezy białek, glikogenu, cholesterolu i NEFA (co redukuje
zużycie ATP i ułatwia utrzymanie równowagi energetycznej serca).
Na mechanizm aktywacji przemian glukozy i NEFA przez AMPK składają się: [13,14]:
1. zwiększone przemieszczanie się na powierzchnię błony komórkowej zarówno transporterów glukozy (GLUT 4) jak i NEFA (FAT/CD36), co przyspiesza dokomórkowy transport
obu tych substratów;
2. pośrednia aktywacja fosfofruktokinazy, co skutkuje przyspieszeniem glikolizy i zwiększoną
produkcją ATP;
3. inaktywacja karboksylazy acetylo-CoA (ACC), co skutkuje kolejno: mniejszą produkcją
malonylo-CoA, mniejszym hamowaniem przez malonylo-CoA transferazy palmitynokarnitynowej I (CPT I, enzym regulujący transport NEFA do mitochondriów, ryc. 4.4.),
przyspieszonym transportem NEFA do mitochondriów oraz ich zwiększonym spalaniem
mitochondrialnym i zwiększoną produkcją ATP.
Eliminacja AMPK zwalnia proces dostosowywania się sercowej produkcji ATP do zwiększonego obciążenia i tym samym zmniejsza rezerwę skurczową miokardium. Zwraca uwagę
fakt, że insulina i AMPK mają podobne punkty uchwytu komórkowego działania, podobnie
wpływają na transport dokomórkowy glukozy i NEFA i glikolizę, i że te efekty metaboliczne
insuliny i aktywacji AMPK są od siebie niezależne.
Ostatecznie, pod wpływem wysiłku fizycznego, ale także pod wpływem stymulacji
współczulnej, rośnie spalanie zarówno węglowodanów (glukoza, kwas mlekowy, glikogen)
jak i NEFA. Jednakże bardziej rośnie spalanie NEFA (bo rośnie poziom NEFA w surowicy,
IV.6). Dodatkowo, odpowiedź zależy od proporcji stężeń we krwi kwasu mlekowego i NEFA.
Im większe jest stężenie kwasu mlekowego tym większe jest jego spalanie kosztem spalania
NEFA [6].
Piśmiennictwo
1. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing
heart. Physiol Rev 2005; 85:1093-1129.
2. Bertrand L, Horman S, Beauloye C, Vanoverschelde JL. Insulin signalling in the heart. Cardiovasc
Res 2008; 79:238-248.
3. Glatz JF, Luiken JJ, Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism:
implications for metabolic disease. Physiol Rev 2010; 90:367-417.
4. Ingwall JS, Weiss RG. Is the failing heart energy starved? on using chemical energy to support cardiac function. Circ Res 2004; 95:135-145.
5. Neubauer S. The failing heart - an engine out of fuel. N Engl J Med 2007; 356:1140-1151.
6. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in
health and disease. Physiol Rev 2010; 90:207-258.
7. Zhang L, Keung W, Samokhvalov V, Wang W, Lopaschuk GD. Role of fatty acid uptake and fatty acid
beta-oxidation in mediating insulin resistance in heart and skeletal muscle. Biochim Biophys Acta
2010; 1801:1-22.
71
8. Lam A, Lopaschuk GD. Anti-anginal effects of partial fatty acid oxidation inhibitors. Curr Opin
Pharmacol 2007; 7:179-185.
9. Lee L, Horowitz J, Frenneaux M. Metabolic manipulation in ischaemic heart disease, a novel approach to treatment. Eur Heart J 2004; 25:634-641.
10. Mjos OD. Effect of free fatty acids on myocardial function and oxygen consumption in intact dogs.
J Clin Invest 1971; 50:1386-1389.
11. Stanley WC, Lopaschuk GD, Hall JL, Mccormack JG. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res 1997; 33:243-257.
12. Stanley WC, Sabbah HN. Metabolic therapy for ischemic heart disease: the rationale for inhibition
of fatty acid oxidation. Heart Fail Rev 2005; 10:275-279.
13. Li C, Keaney JF, Jr. AMP-activated protein kinase: a stress-responsive kinase with implications for
cardiovascular disease. Curr Opin Pharmacol 2010; 10:111-115.
14. Viollet B, Athea Y, Mounier R et al. AMPK: Lessons from transgenic and knockout animals. Front
Biosci 2009; 14:19-44.
72
V. Patofizjologia niedokrwienia i reperfuzji
Andrzej BERĘSEWICZ
V.1. Definicje i charakterystyka ogólna
Produkcja ATP i fosfokreatyny, a następnie czynność skurczowa mięśnia sercowego,
w sposób krytyczny zależą od równowagi między aktualnym przepływem wieńcowym i zapotrzebowaniem na przepływ.
W patologii ludzkiej występują liczne stany (tab. 5.1.), w których dochodzi do nierówności:
Przepływ wieńcowy < Zapotrzebowanie na przepływ
Wszystkie one są definiowane jako niedokrwienie mięśnia sercowego gdyż towarzyszy
im uwalnianie z serca kwasu mlekowego (produkt glikolizy beztlenowej) oraz adenozyny
(produkt postępującego rozkładu nukleotydów adeninowych). Jeżeli deficyt przepływu jest
znaczny i trwa odpowiednio długo, niedokrwienie skutkuje różnymi postaciami uszkodzenia
mięśnia sercowego.
Tab. 5.1. Kliniczne postaci niedokrwienia
Niedokrwienia spoczynkowe
Zawał serca
Spazm naczyniowy
Niedokrwienia około-proceduralne
– plastyka wieńcowa
– krążenie pozaustrojowe
– transplantacja serca
Niedokrwienia wysiłkowe
Angina wysiłkowa
Próba wysiłkowa
Kliniczne postaci niedokrwienia (tab. 5.1.) kończą się obecnie najczęściej spontanicznym
bądź indukowanym powrotem równowagi między przepływem wieńcowym i zapotrzebowaniem na przepływ, czyli reperfuzją (vide koncepcja niedokrwienia spoczynkowego i wysiłkowego, rozdz. V.2). Reperfuzja jest jedynym znanym sposobem ratowania niedokrwionych
73
komórek, ale jest równocześnie czynnikiem uszkadzającym. Ostateczny wynik reperfuzji jest
zawsze sumą jej korzystnych efektów i efektów tzw. reperfuzyjnego uszkodzenie serca.
Uszkodzenia niedokrwienne i reperfuzyjne „w czystej postaci” dają się badać tylko w warunkach eksperymentalnych. Obraz kliniczny różnych postaci niedokrwienia jest z reguły
mieszaniną efektów samego niedokrwienia i następującej po nim reperfuzji. Dlatego w kontekście klinicznym mówimy o uszkodzeniu niedokrwienno-reperfuzyjnym (ang. ischemia/
reperfusion injury) (ryc. 5.1.).
Ryc. 5.1. Koncepcja niedokrwienno-reperfuzyjnego uszkodzenia serca. Zarówno niedokrwienie jak
i reperfuzja są źródłem uszkodzenia. W klinice efekty te sumują się. Długość niedokrwienia jest główną
modyfikowalną determinantą niedokrwiennego uszkodzenia (i wtórnie uszkodzenia reperfuzyjnego).
Kardioprotekcja skutkuje redukcją jedynie reperfuzyjnego uszkodzenia.
Nasilenie uszkodzenia niedokrwiennego jest wypadkową stopnia ograniczenia perfuzji
wieńcowej oraz czasu trwania niedokrwienia:
Uszkodzenie niedokrwienne = % ograniczenia przepływu x długość niedokrwienia
Dodatkowa komplikacja wynika z faktu, że uszkodzenie reperfuzyjne jest tym większe im
większe jest poprzedzające uszkodzenie niedokrwienne. Oznacza to, że obie postaci uszkodzenia zależą ostatecznie od czasu trwania niedokrwienia i stopnia ograniczenia perfuzji
wieńcowej.
W obrazie kliniczno-patofizjologicznym niedokrwienia dominują następujące elementy:
1. Ból wieńcowy;
2. Zaburzenia kurczliwości miokardium;
3. Zaburzenia rytmu;
4. Utrata równowagi energetycznej miokardium skutkująca rozkładem ATP do AMP i adenozyny, które stają się substancjami sygnałowymi. Adenozyna indukuje ból wieńcowy
i powoduje rozszerzenie naczyń. AMP, poprzez kinazę AMP, przekierowuje metabolizm
energetyczny komórek na produkcję ATP z glukozy;
5. Od pewnego etapu niedokrwienia miokardium ulega martwicy, która „wędruje” od wsierdzia do nasierdzia;
74
6. Równocześnie niedokrwienie skutkuje zaburzeniami, które są substratem dla patologicznych procesów uruchamianych w kardiomiocytach (ale także w naczyniach mikrokrążenia w obszarze niedokrwienia) dopiero w reperfuzji. Najważniejsze z nich to: (i) niskie
komórkowe poziomy ADP i AMP i wysokie poziomy fosforu nieorganicznego; (ii) zmiany w mitochondriach usposabiające je do reperfuzyjnej nadprodukcji reaktywnych form
tlenu oraz (iii) kwasica komórkowa skutkująca komórkową akumulacją Ca2+. Kulminacją
tych procesów jest, między innymi, reperfuzyjna aktywacja megakanału mitochondrialnego, ostatecznie skutkująca reperfuzyjną śmiercią komórek (rozdz. V.8.4.). Dodatkową
konsekwencją jest akumulacja i aktywacja komórek zapalnych w obszarze niedokrwionym/
reperfundowanym i związana z tym toksyczność.
Na efekty reperfuzji składają się:
1. Częściowa odbudowa czynności skurczowej miokardium i/lub ochrona kardiomiocytów
przed śmiercią spowodowaną niedokrwieniem. Jest to działanie korzystne i reperfuzja jest
jedynym znanym sposobem ochrony niedokrwionych komórek;
2. Reperfuzyjne uszkodzenie serca – jego istnienia dowodzi fakt, że różne interwencje kardioprotekcyjne zastosowane tuż przed, lub tuż po rozpoczęciu reperfuzji zmniejszają wielkość
zawału u zwierząt eksperymentalnych i ludzi. Tradycyjnie wyróżnia się cztery typy zaburzeń spowodowanych reperfuzją:
(i) Ogłuszenie mięśnia sercowego;
(ii) Reperfuzyjne zaburzenia rytmu;
(iii) Śmierć komórek w wyniku nekrozy i apoptozy. Reperfuzja przyspiesza śmierć komórek
nieodwracalnie uszkodzonych w trakcie niedokrwienia, oraz powoduje śmierć komórek, które w momencie rozpoczęcia reperfuzji były uszkodzone jeszcze w sposób potencjalnie odwracalny;
(iv) Reperfuzyjne uszkodzenie śródbłonka, którego konsekwencjami są:
• akumulacja komórek zapalnych w reperfundowanym obszarze i toksyczność komórkowa z tym związana oraz
• No-reflow phenomenon – stan, w którym, w wyniku zabiegu reperfuzyjnego doszło do
embolizacji naczyń mikrokrążenia, co ogranicza spodziewany powrót perfuzji miokardium i korzyści reperfuzji.
Terminem kardioprotekcja określane są różne postępowania lecznicze mające na celu
ograniczanie uszkodzenia niedokrwienno/reperfuzyjnego miokardium (rozdz. VI). Kardioprotekcja może być wymierzona przeciwko uszkodzeniu niedokrwiennemu albo reperfuzyjnemu. Jedyny skuteczny obecnie sposób ograniczania niedokrwiennego uszkodzenia polega
na skracaniu czasu do reperfuzji (skracaniu długości niedokrwienia), co pośrednio skutkuje
także mniejszym uszkodzeniem reperfuzyjnym. Leczenie kardioprotekcyjne, jeżeli zmniejsza
uszkodzenie niedokrwienno/reperfuzyjne, działa poprzez redukcję reperfuzyjnej komponenty uszkodzenia.
Budzącym nadzieje kliniczne, ale ciągle słabo poznanym aspektem patofizjologii niedokrwienia/reperfuzji są endogenne mechanizmy kardioprotekcyjne. Najlepiej poznane i najbliższe wykorzystania terapeutycznego jest tzw. „hartowanie” mięśnia sercowego. Jest to mechanizm polegający na tym, że krótkie nieuszkadzające incydenty niedokrwienia/reperfuzji
aktywują endogenne mechanizmy ochronne miokardium (aktywacja szlaków sygnalizacyj75
nych i/lub genów o działaniu kardioprotekcyjnym) i w ten sposób uodparniają miokardium
na toksyczne działanie ewentualnych kolejnych niedokrwień (rozdz. VI.3).
V.2. Dwa mechanizmy niedokrwienia
Ze względu na mechanizm, wyróżnia się dwa typy niedokrwienia, określane w literaturze
anglosaskiej jako supply ischemia (konsekwencja ograniczenia lub zatrzymania spoczynkowej
perfuzji wieńcowej) i demand-induced ischemia (konsekwencja niewystarczającego wzrostu
przepływu wieńcowego w czasie wysiłku). W tym opracowaniu, terminy te są tłumaczone
jako odpowiednio „niedokrwienie spoczynkowe” i „niedokrwienie wysiłkowe”. Termin
„niedokrwienie wysiłkowe” odnosi się także do zaburzeń wywołanych innymi niż wysiłek
fizyczny czynnikami, zwiększającymi obciążenie serca (szybka stymulacja serca, infuzja dobutaminy, aktywacja współczulna etc.). Obecna wiedza na temat patofizjologii niedokrwienia
i reperfuzji mięśnia sercowego pochodzi głównie z badań na zwierzęcych modelach niedokrwienia spoczynkowego (najczęściej izolowane serca szczura z ograniczonym lub zatrzymanym spoczynkowym przepływem wieńcowym lub serca in situ psa lub świni z podwiązaną
tętnicą wieńcową). Fizjologia niedokrwienia wysiłkowego jest bardzo słabo zbadana. Nie jest,
więc pewne czy różni się ona od fizjologii niedokrwienia spoczynkowego jedynie ilościowo
czy również jakościowo.
V.2.1. Niedokrwienie spoczynkowe
Prototypami niedokrwienia spoczynkowego są zawał serca i niestabilna choroba wieńcowa (ale także spazm naczyniowy, krążenie pozaustrojowe, plastyka wieńcowa). W stanach
tych niedokrwienie jest wynikiem zatrzymania lub ograniczenia spoczynkowej perfuzji
wieńcowej, najczęściej w wyniku zwężenia lub zamknięcia tętnicy wieńcowej.
Krążenia wieńcowe świni, szczura i królika są całkowicie pozbawione krążenia obocznego. Dlatego niedrożność tętnicy nasierdziowej skutkuje u tych zwierząt całkowitym zatrzymaniem perfuzji wieńcowej w dorzeczu niedrożnej tętnicy i powstaniem transmuralnego zawału
serca po ~30 minutach niedokrwienia. U ludzi (ale także u psa i kota) wieńcowe krążenie
oboczne jest dość dobrze rozwinięte. Dlatego całkowite zamknięcie tętnicy wieńcowej skutkuje zwykle jedynie częściowym i niehomogennym spadkiem perfuzji w dorzeczu niedrożnej
tętnicy (ryc. 5.2.D). Spadek ten jest większy w warstwie podwsierdziowej niż podnasierdziowej ściany komory, a także większy w centrum zawału niż na jego obrzeżach. Towarzyszy
mu, proporcjonalny do stopnia ograniczenia perfuzji, spadek tlenowej produkcji ATP, wzrost
produkcji kwasu mlekowego, kwasica komórkowa, spadek kurczliwości mięśnia sercowego,
a z czasem postępująca śmierć kardiomiocytów. Przestrzenne nasilenie tych zmian jest odwzorowaniem niehomogenności perfuzji.
V.2.2. Niedokrwienie wysiłkowe
Niedokrwienie wysiłkowe występuje powszechnie u osób ze stenozą tętnicy wieńcowej
i stabilną chorobą wieńcową i jest statystycznie najczęstszym typem niedokrwienia u ludzi.
70–80% epizodów takiego niedokrwienia przebiega bezbólowo (silent ischemia) i są one identyfikowane dopiero na podstawie badania EKG [1].
76
Ryc. 5.2. Wpływ wysiłku fizycznego na perfuzję wieńcową w różnych warstwach ściany lewej komory
psów w obszarze za zwężeniem tętnicy nasierdziowej. W spoczynku przepływ w warstwie podwsierdziowej (Endo) jest nieco większy niż w podnasierdziowej (Epi) (stosunek przepływów Endo/Epi ~1,2).
(A) Kontrola – wysiłek fizyczny powoduje podobny wzrost przepływu w Epi i Endo; (B) Stenozą 40% – wysiłek powoduje normalny wzrost przepływu w Epi i jedynie niewielki wzrost przepływu w Endo (Endo/Epi
<1) i wobec tego warstwa ta ma deficyt przepływu w stosunku do potrzeb; (C) Stenoza 60% – wysiłek powoduje mniejszy niż normalnie wzrost przepływu w Epi oraz jego spadek, w stosunku do przepływu spoczynkowego, w Endo i wobec tego deficyt przepływu dotyczy Endo i Epi; (D) Transmuralna dystrybucja
perfuzji wieńcowej w obszarze serca objętym zawałem. We wszystkich warstwach perfuzja jest mniejsza od
kontrolnej, ale ograniczenie perfuzji jest szczególnie duże w Endo. Dane w A, B i C pochodzą z ref . [2]
Stenozy zmniejszające średnicę tętnicy nasierdziowej nawet o ~85% pozostają bez wpływu na spoczynkowy przepływ wieńcowy. Przepływ ten ograniczają dopiero stenozy >85%
(tzw. stenozy krytyczne) (rozdz. III.5). Zwężenia tętnicy w zakresie 50–85% (tzw. stenozy subkrytyczne) ograniczają natomiast rezerwę wieńcową [2]. Dlatego obszary serca zaopatrywane przez tętnice ze stenozą subkrytyczną mają niedobory perfuzji wieńcowej dopiero w momencie zwiększonego obciążenia serca (np. wysiłek fizyczny, aktywacja współczulna, infuzja
dobutaminy, szybka czynność serca). Następuje wtedy, podyktowany obciążeniem, wzrost
perfuzji wieńcowej. Jednakże w obszarze za stenozą, zwłaszcza w warstwie podwsierdziowej
ściany komory, jest on mniejszy niż by tego wymagało aktualne obciążenie (bo rezerwa wieńcowa jest ograniczona) (ryc. 5.2.). W efekcie dochodzi do niedokrwienia, w czasie którego
aktualny przepływ wieńcowy może być nawet większy niż przepływ spoczynkowy. Jest to
na ogół krótkotrwała i łagodna (w porównaniu z zawałem) postać niedokrwienia, w której
mięsień sercowy jest perfundowany utlenowaną krwią w objętości, która jest bliska objętości
spoczynkowej (ale zbyt mała w stosunku do potrzeb). W efekcie produkcja energii odbywa się
ciągle w mechanizmie tlenowych, ale jej wielkość nie nadąża za potrzebami.
Pierwszy kompletny opis wydarzeń towarzyszących niedokrwieniu wysiłkowemu pochodzi sprzed czterdziestu lat [3,4]. Badanie objęło osoby ze stabilną chorobą wieńcową,
anginą wysiłkową w wywiadzie, normalnym zapisem EKG i prawidłową frakcją wyrzucania lewej komory. U większości badanych osób, już w 1–2 minucie wysiłku fizycznego lub
szybkiej stymulacji komór dochodziło do zwiększonego wypływu kwasu mlekowego z zatoki wieńcowej i netto sercowej produkcji kwasu mlekowego (marker glikolizy beztlenowej
w sercu). Z pewnym opóźnieniem obserwowano postępujące obniżenie odcinka ST elektro77
kardiogramu i dopiero w następnej kolejności pojawiał się ból wieńcowy i zaburzenia czynności hemodynamicznej serca w postaci wzrostu ciśnienia końcoworozkurczowego w lewej
komorze. Opisane zmiany ustępowały w ciągu kilku minut po normalizacji obciążenia. Te
wczesne obserwacje znalazły potwierdzenie w późniejszych badaniach u ludzi i zwierząt eksperymentalnych [2].
Podsumowując, bezpośrednie pomiary perfuzji wieńcowej u zwierząt eksperymentalnych
i ludzi [2,5-8] ze zwężeniami tętnic wieńcowych potwierdzają opinię, że niedokrwienie wysiłkowe mięśnia sercowego u pacjentów ze stabilną chorobą wieńcową jest skutkiem raczej zbyt
małego (w stosunku do aktualnego obciążenia) wzrostu przepływu wieńcowego, niż ograniczenia przepływu poniżej wartości spoczynkowych. Efektem tego nienadążania przepływu
za obciążeniem jest stan nierównowagi między podażą a popytem na przepływ, spełniający
kryteria definicji niedokrwienia, w tym wypadku niedokrwienia wysiłkowego. W przypadku
obszarów serca z dużą stenozą i wobec tego niewielką rezerwą wieńcową, zwiększone obciążenie serca może skutkować, zwłaszcza w warstwie podwsierdziowej, ograniczeniem przepływu
poniżej jego wartości spoczynkowej, co należałoby klasyfikować jako łagodne niedokrwienie
o typie niedokrwienia spoczynkowego (supply ischemia) (ryc. 5.2.C). Jest prawdopodobne
wobec tego, że w obszarach serca zaopatrywanych przez zwężoną subkrytycznie tętnice wieńcową, wysiłek fizyczny i inne czynniki obciążające mogą prowokować równocześnie niedokrwienie wysiłkowe (demand induced ischemia) w warstwie podnasierdziowej i niedokrwienie spoczynkowe (supply ischemia) w warstwie podwsierdziowej. Nie jest pewne, czy oba
te typy niedokrwienia, czy tylko niedokrwienie o typie supply ischemia, jest odpowiedzialne
za symptomatologię stabilnej choroby wieńcowej (produkcja kwasu mlekowego, zaburzenia
kurczliwości, zmiany odcinka ST elektrokardiogramu oraz ból wieńcowy etc.).
V.3. Degradacja ATP w niedokrwieniu – regulacyjne i niekorzystne
konsekwencje
Niezależnie od wielkości i rodzaju niedokrwienia, produkcja ATP w ognisku niedokrwienia nie nadąża za jego rozkładem.
Skutkuje to (ryc. 5.3.): (i) postępującą degradacją ATP – kolejno do ADP, AMP i adenozyny; (ii) przechodzeniem adenozyny do przestrzeni pozakomórkowej przy udziale błonowego transportera oraz (iii) dalszą degradacją adenozyny w przestrzeni pozakomórkowej do
inozyny, hipoksantyny, ksantyny i kwasu moczowego. Zaburzenia te mają natychmiastowe
konsekwencje dla niedokrwionego miokardium (ryc. 5.4.) oraz konsekwencje odległe ujawniające się dopiero w czasie reperfuzji.
Niedobór ATP skutkuje szybką utratą kurczliwości miokardium. Równocześnie zmiany
stężeń ATP i jego metabolitów uruchamiają wymienione poniżej ścieżki regulacyjne:
1. Obniżenie komórkowego stężenia ATP jest czynnikiem aktywującym błonowe kanały potasowe, tzw. kanały potasowe ATP-zależne (kanały KATP). Skutkuje to wypływem jonów K+
z niedokrwionych kardiomiocytów, ich depolaryzacją i przepływem prądu „uszkodzenia”
(injury current) od obszaru niedokrwionego do prawidłowo ukrwionego. W badaniu EKG
jest to „widoczne” jako zmiany w odcinku ST-T. Jest także czynnikiem prowokującym groźne zaburzenia rytmu towarzyszące pierwszym minutom niedokrwienia;
78
Ryc. 5.3. Szlaki degradacji i resyntezy ATP w niedokrwieniu. Komórkowe stężenia ATP maleją a AMP
i fosforu nieorganicznego (Pi) rosną. Rośnie także zewnątrzkomórkowe stężenie adenozyny. Zmiany
mają działania regulacyjne (vide ryc. 5.4.). Adenozyna wraz z jej metabolitami jest wypłukiwana z serca,
zwłaszcza w reperfuzji, i nie może stanowić podstawy do resyntezy ATP. T – transporter błonowy adenozyny, blokowany przez dipirydamol.
Ryc. 5.4. Natychmiastowe konsekwencje niedokrwiennej degradacji ATP i gromadzenia się produktów
jego rozpadu. Szczegóły w tekście
2. Wzrost komórkowego stężenia AMP jest czynnikiem stymulującym kinazę AMP (AMPK)
(rozdz. IV.7.), która poprzez fosforylację kluczowych enzymów zaangażowanych w metabolizm energetyczny glukozy i NEFA, przekierowuje metabolizmu niedokrwionego mięśnia sercowego z przemian NEFA na przemiany glukozy;
79
Tabela 5.2. Efekty stymulacji receptorów adenozynowych A1 i A2 w sercu
Typ komórek
Węzeł zatokowy
Węzeł A-V
Mięsień przedsionka
Mięsień komorowy
Naczynia
– komórki śródbłonka
– komórki mięśni gładkich
Okołonaczyniowe nerwy współczulne
– eferentne
– aferentne
Receptor
Hamowanie produkcji cAMP, zwalnianie
A1
rytmu zatokowego
A1
Hamowanie produkcji cAMP, zwalnianie
przewodzenia
A1
Skracanie potencjału czynnościowego
i okresu refrakcji
A1
Przeciwdziałanie efektom aktywacji
β-receptorów adrenergicznych. Aktywacja
mechanizmu hartowania niedokrwieniem
A2
A2
Aktywacja produkcji cAMP, rozkurcz naczyń
Aktywacja produkcji cAMP, rozkurcz naczyń
A1
A1
Hamowanie uwalniania noradrenaliny
Aferentna aktywacja skutkująca percepcją
bólu
3. Gromadząca się w przestrzeni pozakomórkowej adenozyna jest substancją czynną działającą poprzez aktywację błonowych receptorów A1 i A2 zlokalizowanych na różnych komórkach w sercu (tab. 5.2.). Natychmiastowymi efektami działania adenozyny są, między
innymi, rozkurcz naczyń i aktywacja okołonaczyniowych włókien współczulnych wyzwalająca ból wieńcowy (rozdz. V.4.). Adenozyna jest także mediatorem wyzwalającym zjawisko hartowania niedokrwieniem (rozdz. VI.3).
Odległą konsekwencją postępującej degradacji ATP jest wypływ jego metabolitów z serca. W postaciach niedokrwienia z zachowanym przepływem, wypływ ten ma miejsce przez
cały czas niedokrwienia. W przypadku zawału, metabolity ATP (adenozyna, inozyna, hipoksantyna etc.) gromadzą się w przestrzeni zewnątrzkomórkowej i są intensywnie wypłukiwane
z miokardium dopiero w czasie reperfuzji. Jest to jeden z tzw. paradoksów reperfuzji, którego
szkodliwe konsekwencje są dwojakie. (i) Reperfuzja, której celem jest przywrócenie tlenowej
produkcji ATP, paradoksalnie utrudnia ten proces poprzez pozbawianie miokardium substratu do resyntezy ATP; (ii) Deficyt komórkowy ATP i ADP i wzrost komórkowego poziomu
fosforu nieorganicznego sprzyja toksycznej aktywacji megakanału mitochondrialnego (rozdz.
V.8.4). Zgodnie z tym rozumowaniem, reperfuzja miokardium z prekursorami syntezy ATP
(np. z adenozyną lub prekursorami jej syntezy) przyspiesza odbudowę czynności skurczowej
miokardium w czasie reperfuzji (eksperymenty zwierzęce i krążenie pozaustrojowe u ludzi).
V.4. Ból wieńcowy (angina pectoris)
Ból w klatce piersiowej (angina pectoris) jest częstym zjawiskiem w niedokrwieniu mięśnia sercowego. Jego analiza jest jednak mało przydatna w określaniu obecności, lokalizacji
i wielkości niedokrwienia miokardium. Wynika to z następujących faktów:
• Badania u ambulatoryjnych pacjentów z chorobą wieńcową wykazały, że około 75% epizodów niedokrwienia stwierdzanych w dobowym badaniu EKG ma przebieg bezbólowy
80
(nieme niedokrwienie) a dwie trzecie incydentów bólowych w klatce piersiowej przebiega
bez cech niedokrwienia w EKG [1,9];
• Epizody niedokrwienia trwające <3 min pozostają zazwyczaj nieme. Natomiast dłuższe
i bardziej nasilone niedokrwienia mogą mieć przebieg zarówno bólowy jak i bezbólowy,
nawet u tego samego pacjenta [10];
• Lokalizacja bólu wieńcowego w obrębie klatki piersiowej i/lub poza nią nie wykazuje korelacji z lokalizacją obszaru niedokrwienia w obrębie serca. Podobnie, infuzja adenozyny
(czynnik wyzwalający ból wieńcowy, vide poniżej) do prawej i lewej tętnicy wieńcowej skutkowała podobną lokalizacją bólu wieńcowego u 75% badanych pacjentów [10];
• Brak jest wyraźnej czasowej korelacji między początkiem niedokrwienia i pojawianiem się
bólu wieńcowego. U pacjentów z anginą wysiłkową (łagodna postać niedokrwienia) a także u pacjentów z kardiologicznym zespołem X, dolegliwości bólowe często pojawiają się
wcześniej niż niedokrwienne zmiany w EKG. Natomiast u pacjentów z zaawansowanym
niedokrwieniem, spowodowanym spazmem naczyniowym, najwcześniejszą konsekwencją
niedokrwienia były lokalne zaburzenie kurczliwości mięśnia sercowego a ból wieńcowy występował dopiero z paruminutowym opóźnieniem [10];
• Kobiety vs. mężczyźni; kobiety częściej niż mężczyźni mają dolegliwości w klatce piersiowej
o typie dolegliwości wieńcowych. Z drugiej strony, w grupie kobiet i mężczyzn z podobnym
zawansowaniu dolegliwości wieńcowych, u kobiet zaawansowanie arteriograficznych zmian
miażdżycowych jest mniejsze natomiast natężenie zmian niedokrwiennych w dobowym
EKG większe [11].
Szlak nerwowy zapewniający transmisję bodźców bólowych z miokardium do kory mózgowej składa się z następujących elementów [10,12].
1. Czynnikiem wyzwalającym ból jest adenozyna działająca poprzez receptory A1 na okołonaczyniowych aferentnych współczulnych włóknach nerwowych (i być może przywspółczulnych) przebiegających równolegle do odgałęzień tętnic wieńcowych;
2. Włókna te wstępują do zwojów sympatycznych (C7-T4) a następnie tworzą synapsę z komórkami nerwowymi w rogach grzbietowych rdzenia kręgowego. Tam spotykają się i nawzajem na siebie wpływają bodźce czuciowe z serca i z innych części ciała. Dodatkowo,
komórki te podlegają modulacji przez impulsy pochodzące z wyższych partii układu nerwowego, co razem wziąwszy stanowi anatomiczną podstawę promieniowania bólu wieńcowego w różne okolice ciała a także skuteczności stymulacji rdzenia kręgowego w leczeniu
lekoopornej anginy [13];
3. Impulsy są dalej przekazywane szlakami rdzeniowo-wzgórzowymi do tylnego podwzgórza,
gdzie podlegają dalszej modulacji;
4. Percepcja bólu związana jest dopiero z przekazywaniem impulsów do kory mózgowej;
5. Dominuje obecnie przekonanie, że zmienna percepcja bólu wieńcowego, typowa dla choroby wieńcowej, jest wypadkową lokalnego działania adenozyny i modulacji sygnału adenozynowego na poziomie najpierw zwojów współczulnych a następnie rdzenia kręgowego
i wyższych partii centralnego układu nerwowego.
Następujące argumenty przemawiają na rzecz hipotezy, że adenozyna jest aktywatorem
receptorów bólowych w sercu i czynnikiem wyzwalającym dolegliwości wieńcowe: [10]
1. Adenozyna powstaje w i jest uwalniana z niedokrwionego miokardium;
81
2. Adenozyna podana dowieńcowo powoduje typowy ból anginalny, także u zdrowych osób
i nie towarzyszą temu objawy niedokrwienia. Dipirydamol (bloker komórkowego wychwytu adenozyny) potęguje, a teofilina (nieselektywny bloker receptorów A1 i A2) i bamifilina
(selektywny bloker receptorów A1) hamują ten efekt;
3. Teofilina i bamifilina zmniejszają dolegliwości bólowe w wysiłkowej anginie, mimo że nie
wpływają na wielkość niedokrwienia;
4. Adenozyna podana śródskórnie wywołuje ból, który jest blokowany przez bamifilinę;
5. Ból wywołany adenozyną nie jest wtórny do rozkurczu naczyń i ewentualnej mechanicznej
aktywacji okołonaczyniowych zakończeń nerwowych. Niemniej jednak, mechaniczne rozciągniecie tętnicy wieńcowej (balon, stent), samo przez się, jest czynnikiem indukującym
ból. Może to tłumaczyć występowanie bólu w klatce piersiowej u osób z implantowanym
stentem tętnicy wieńcowej pod nieobecność niedokrwienia mięśnia sercowego;
6. Nasierdziowa aplikacja adenozyny lub selektywnego agonisty receptorów A1 skutkuje aktywacją sercowych aferentnych nerwów współczulnych. Agonista receptorów A2 nie ma
takiego działania.
Niektórzy pacjenci nigdy nie odczuwają bólu wieńcowego a u innych występują niedokrwienia i bólowe, i bezbólowe. Ma to prawdopodobnie związek z upośledzeniem ogólnej,
a nie tylko sercowej, percepcji bólu u tych osobników. Wykazano, że ogólna percepcja bólu
jest obniżona, a występowanie niemych niedokrwień serca jest zwiększone, u: (i) osób z nadciśnieniem tętniczym; (ii) mężczyzn w porównaniu z kobietami oraz (iii) przedstawicieli rasy
kaukaskiej w porównaniu z czarną. Natomiast depresja oraz niski status społeczny są czynnikami obniżającymi ogólny próg percepcji bólu i zwiększającymi występowanie bólowych
incydentów niedokrwienia [14].
Liczne badania sugerują, że percepcja bólu wieńcowego jest poważnie zwiększona u pacjentów z kardiologicznym zespołem X i że ma to związek z przynajmniej dwoma zaburzeniami transmisji bodźców bólowych z serca do kory. Po pierwsze, wykazano zaburzenia w sercowych włóknach współczulnych objawiające się upośledzonym wychwytywaniem przez nie
noradrenaliny [15]. Po drugie wykazano brak tzw. habituacji bodźców czuciowych w zespole X, co może tłumaczyć występowanie przedłużających się incydentów anginy wysiłkowej
w tym zespole [12].
Występowanie niemych niedokrwień i niemych zawałów jest szczególnie częste w cukrzycy. Ma to prawdopodobnie związek z neuropatią sercowych włókien współczulnych,
obecność której potwierdzono w badaniach autopsyjnych osób z cukrzycą zmarłych w wyniku niemego niedokrwienia [10].
V.5. Zaburzenia kurczliwości mięśnia sercowego w niedokrwieniu
i reperfuzji
1. Każdej postaci niedokrwienia towarzyszą natychmiastowe (w ciągu sekund) zaburzenia kurczliwości miokardium. W niedokrwieniu wysiłkowym polegają one na braku wysiłkowego wzrostu
kurczliwości, a w niedokrwieniu spoczynkowym – na bezwzględnym spadku kurczliwości;
2. Wczesne zaburzenia kurczliwości wyprzedzają w czasie spadek komórkowych poziomów ATP
i są reakcją obronną, w której komórki „wyłączają” energochłonny skurcz a zaoszczędzony
82
ATP przeznaczają na podstawowe procesy życiowe. Dalszy przebieg wydarzeń zależy od wielkości niedokrwienia;
3. W obszarach z umiarkowanie ograniczoną spoczynkową perfuzją wieńcową (<50%), kurczliwość maleje proporcjonalnie do ograniczenia perfuzji, a zasoby ATP i CrP, po przejściowym
spadku, normalizują się. Ten stan, znany jako hibernacja miokardium (obniżony przepływ
i kurczliwość i zachowany bilans energetyczny) może trwać kilkadziesiąt godzin zanim komórki zaczną umierać;
4. W obszarach ze znacznie ograniczoną perfuzją (>50%) skurcz ustaje, zasoby ATP i CrP szybko się wyczerpują i komórki stopniowo umierają;
5. Jeżeli niedokrwienie jest niewielkie lub trwa na tyle krótko, że komórki jeszcze nie umarły,
reperfuzja całkowicie odbudowuje skurcz, ale jest to wielo-godzinny/-dniowy/-tygodniowy
proces, znany jako ogłuszenie mięśnia sercowego. Mechanizm ogłuszenia „ostrego” polega na
reperfuzyjnym Ca2+-zależnym enzymatycznym trawieniu białek kurczliwych i na zmianach
obniżających efektywność mechaniczną mięśnia. Mechanizm ogłuszenia „przewlekłego”
obejmuje zmniejszoną ekspresję białek kurczliwych i zanik aparatu kurczliwego;.
6. Lokalne odwracalne zaburzenia kurczliwości lewej komory u osób z CNS (obszary hypo-,
a- i dyskinetyczne, reagujące zmianą kurczliwości na dobutaminę i odzyskujące kurczliwość
w wyniku plastyki wieńcowej lub CABG) są postacią „przewlekłego” ogłuszenia, spowodowanego powtarzającymi się incydentami wysiłkowego niedokrwienia i reperfuzji, w obszarach
serca zaopatrywanych przez subkrytycznie zwężoną tętnicę wieńcową.
V.5.1. Kurczliwość w niedokrwieniu wysiłkowym; próba dobutaminowa
Brak jest systematycznych badań klinicznych, dotyczących wpływu niedokrwienia wysiłkowego na kurczliwość różnych warstw miokardium, zwłaszcza w konfrontacji z pomiarami
perfuzji wieńcowej. Dostępne są jedynie fragmentaryczne dane, głównie eksperymentalne.
Dla przykładu, u psów ze stenozą wieńcową tak dobraną by obszar za stenozą miał normalny
spoczynkowy przepływ wieńcowy, ale był pozbawiony rezerwy wieńcowej, wysiłek fizyczny
powodował 200% wzrost przepływu w obszarze kontrolnym, a w obszarze za stenozą 100%
wzrost przepływu w warstwie podnasierdziowej i 50% spadek przepływu w warstwie podwsierdziowej oraz 70% spadek kurczliwości całego obszaru (pomiar echo przyrostu grubości
ściany lewej komory w czasie systole) [16]. W podobnym badaniu u psów z subkrytyczną
stenozą, dobutamina powodowała wzrost przepływu w warstwie podnasierdziowej i środkowej a w warstwie podwsierdziowej przepływ pozostawał na spoczynkowym poziomie. Mimo
tylko podwsierdziowego deficytu perfuzji, dobutamina skutkowała spadkiem kurczliwości
segmentu miokardium zaopatrywanego przez stenotyczną tetnicę (echo) [17].
Dość dobrze został przebadany wpływ wzrastających dawek dobutaminy (wzrastającego
obciążenia) na przepływ wieńcowy i kurczliwość obszarów serca za stenozą. Dla przykładu, w eksperymentach Calnon’a i wsp. [18] podawano wzrastające dawki dobutaminy psom
kontrolnym, oraz z łagodną (niewielkie ograniczenie rezerwy wieńcowej) i zaawansowaną
subkrytyczną stenozą wieńcową (submaksymalne ograniczenie rezerwy). U zwierząt kontrolnych dobutamina powodowała, proporcjonalny do dawki, wzrost prefuzji wieńcowej (scyntygrafia) i kurczliwości miokardium (echo). W obszarach za łagodną stenozą przepływ wzrastał
proporcjonalnie do dawki dobutaminy, ale mniej niż w normalnym sercu i wobec tego de83
ficyt przepływu narastał wraz z dawką dobutaminy. Skutkowało to dwufazowymi zmianami
kurczliwości. Pod wpływem małych dawek dobutaminy kurczliwość rosła jak w normalnych
sercach. Natomiast w obecności większych dawek wzrosty te były coraz mniejsze, a przy najwyższej badanej dawce nie było żadnego wzrostu. W obszarach za zaawansowaną stenozą,
dobutamina nie powodowała żadnych zmian przepływu i proporcjonalny do dawki spadek
kurczliwości (spowodowany, jak można przypuszczać, postępującym niedoborem przepływu
w stosunku do obciążenia).
Podobne dwufazowe zmiany kurczliwości pod wpływem wzrastających dawek dobutaminy [19] oraz testu wysiłkowego [20] obserwowano w obszarach zaopatrywanych przez
zmienione tętnice wieńcowe także u ludzi z chorobą wieńcową.
Sumując, te i inne badania pokazują, że czynniki obciążające mięsień sercowy (wysiłek
fizyczny, dobutamina, szybki rytm, etc.) skutkują:
1. w obszarach zaopatrywanych przez łagodną subkrytyczną stenozę – wzrostem perfuzji
wieńcowej i siły skurczu miokardium, ale wzrosty te są generalnie mniejsze niż w normalnych obszarach serca;
2. w obszarach zaopatrywanych przez „prawie-krytyczną” stenozę – brakiem wzrostu przepływu i kurczliwości lub nawet bezwzględnym spadkiem przepływu i kurczliwości.
V.5.2. Kurczliwość w niedokrwieniu spoczynkowym; hibernowany mięsień sercowy
Osłabienie siły skurczu mięśnia sercowego następuje już w ciągu pierwszych kilku sekund niedokrwienia i jest to najwcześniejszy objaw niedokrwienia. U ludzi w trakcie plastyki
wieńcowej stwierdzono hipokinezę, a następnie dyskinezę niedokrwionego obszaru miokardium już w ~19 sekundzie po inflacji balona powodującej zatrzymanie przepływu w tętnicy
wieńcowej. Kurczliwość normalizowała się w ~17 sekundzie po deflacji balona [21].
Niedokrwiennym zaburzeniom kurczliwości towarzyszy zmniejszenie komórkowych poziomów CrP i początkowo, paradoksalnie, niewielki spadek poziomów ATP. Dlatego mechanizm
zaburzeń kurczliwości we wczesnej fazie niedokrwienia jest niejasny. Niedokrwienie powoduje
szybkie zakwaszenie komórki sercowej, co jest procesem spowodowanym rozkładem ATP oraz
akumulacją kwasu mlekowego. Właśnie kwasica komórkowa jest prawdopodobną przyczyną
wczesnych zaburzeń kurczliwości w niedokrwieniu, gdyż obniża powinowactwo troponiny do
Ca2+, zwiększa wychwytywanie i zmniejsza uwalnianie Ca2+ z siateczki śródplazmatycznej oraz
zmniejsza aktywność błonowych kanałów wapniowych. W późniejszej fazie poważnego niedokrwienia (minuty, godziny) utrata kurczliwości koreluje z utratą komórkowych zasobów ATP
i CrP, a następnie z postępującym nieodwracalnym uszkodzeniem kardiomiocytów [22].
Natychmiastowa utrata kurczliwości kardiomiocytów towarzysząca niedokrwieniu, jest
prawdopodobnie elementem endogennego mechanizmu obronnego serca znanego jako „hibernacja mięśnia sercowego” (hibernating myocardium) [23-26]. Postuluje się, że w mechanizmie tym niedokrwione komórki „wyłączają” energochłonny skurcz (~80% komórkowego
zużycia ATP) a zaoszczędzony ATP „przeznaczają” na podstawowe procesy życiowe. W przypadku całkowitego niedokrwienia hibernacja działa prawdopodobnie jedynie krótko, ale brak
jest badań na ten temat. Natomiast, w obszarach z umiarkowanie ograniczoną spoczynkową
perfuzją wieńcową (<50%), „hibernacja” pozwala przeżyć komórkom kilkunastogodzinne
okresy niedokrwienia.
84
Hibernacja była intensywnie badana w modelu łagodnego, spoczynkowego niedokrwienia, głównie w sercu psa [27] i świni [28]. W eksperymentach tych zakładano przewiązkę na
tętnicy wieńcowej tak, by ograniczenie spoczynkowej perfuzji wieńcowej wynosiło 30–50%.
W tak niedokrwionym obszarze stwierdzono, że:
1. Kurczliwość mięśnia malała w stopniu proporcjonalnym do ograniczenia przepływu wieńcowego i po osiągnięciu nowego, niższego poziomu nie ulegała zmianie przez następnych kilka
godzin eksperymentu. Innymi słowy, w obecności łagodnego, częściowego niedokrwienia
ciągle istniało sprzężenie między przepływem wieńcowym i siłą skurczu (perfusion-contraction matching). Reperfuzja skutkowała bardzo powolną, ale całkowitą, normalizacją siły skurczu miokardium, co oznaczało, że raczej bliżej nieokreślone zaburzenia czynnościowe, a nie
śmierć kardiomiocytów, były przyczyną niedokrwiennych zaburzeń kurczliwości;
2. W pierwszych kilku minutach niedokrwienia obserwowano znaczny spadek komórkowych
poziomów ATP i CrP, które jednak w ciągu następnej godziny niedokrwienia powracały do
normy i pozostawały na normalnym poziomie przez kilka następnych godzin eksperymentu. Od pewnego momentu niedokrwienia, jedyną obserwowaną jego konsekwencją były
zaburzenia kurczliwości, przy w pełni zachowanej równowadze energetycznej;
3. W pierwszych minutach niedokrwienia mięsień sercowy produkował duże ilości kwasu
mlekowego, co jest uznanym wskaźnikiem niedokrwienia. Wraz z upływem czasem, produkcja kwasu mlekowego jednak malała i od 60–90 minuty niedokrwienia mięsień sercowy
był ponownie netto konsumentem kwasu mlekowego.
Mimo licznych badań, komórkowy mechanizm hibernacji jest ciagle nieznany, i nie jest
pewne jak długo może ona chronić miokardium, bez wywoływania degeneracji i/lub śmierci
kardiomiocytów, zwłaszcza w sercu człowieka. Podstawowa wiedza na temat hibernacji pochodzi z badań eksperymentalnych. W różnych modelach zwierzęcych udaje się uzyskać stan
krótkoterminowej hibernacji trwającej kilkanaście godzin. Natomiast próby opracowania modelu zwierzęcego przewlekłej hibernacji (trwającej tygodnie/miesiące) nie powiodły się. Między innymi, dlatego że częściowe zwężenie tętnicy wieńcowej u zwierząt eksperymentalnych
pobudza rozwój krążenia obocznego, co powoduje szybką i całkowitą normalizację spoczynkowego przepływu wieńcowego w obszarze za stenozą [23,28]. Znika wobec tego podstawowe kryterium rozpoznania obszaru hibernowanego, jakim jest ograniczenie spoczynkowego
przepływu wieńcowego. Jednakże, mimo normalizacji spoczynkowego przepływu, obszary za
stenozą mają znacznie ograniczoną rezerwę wieńcową, ciągle słabiej się kurczą, reagują wzrostem kurczliwości na stymulację dobutaminą [23,28,29] i, co istotne, już po 24 godzinach od
częściowego podwiązania tętnicy dochodzi w nich do postępującej ogniskowej degeneracji
kardiomiocytów i ich stopniowego wymierania [30]. Innymi słowy, badania eksperymentalne
potwierdzają obecność w sercu stanu, który można określić jako „hibernacja ostra”, nie potwierdzają natomiast istnienia „hibernacji przewlekłej”. Nie jest pewne, czy i w jakim stopniu
przytoczone dane eksperymentalne odnoszą się do patologii ludzkiej.
V.5.3. Kurczliwość w reperfuzji i ogłuszenie
Obok niedokrwienia, także sama reperfuzja jest źródłem zaburzeń kurczliwości miokardium, określanych jako „ogłuszenie mięśnia sercowego” (stunned myocardium). Ogłuszenie
jest definiowane jako przewlekłe, odwracalne upośledzenie kurczliwości mięśnia sercowego,
85
występujące w reperfundowanym mięśniu, mimo że w mięśniu nie doszło do martwicy komórek i mimo że ma on prawidłową, lub prawie prawidłową, perfuzję wieńcową [31-33]. Podstawą różnicowania hibernacji i ogłuszenia jest fakt, że w hibernacji przepływ wieńcowy jest
aktualnie ograniczony (uszkodzenie niedokrwienne, perfusion-contraction match), a w ogłuszeniu przepływ wieńcowy jest prawidłowy, choć poprzednio był ograniczony (uszkodzenie
reperfuzyjne, perfusion-contraction mismatch) [31,34].
Ogłuszenie po raz pierwszy opisał Heyndrickx i wsp. w 1975 roku [35]. Autorzy ci podwiązywali tętnicę wieńcową u psów i stwierdzili, że niedokrwienia trwające >15 minut powodowały nieodwracalne uszkodzenia miokardium i trwałe upośledzenie kurczliwości,
mimo reperfuzji. Reperfuzja w pełni przywracała kurczliwość niedokrwionego obszaru jedynie w przypadku niedokrwień trwających <15 minut, ale powroty kurczliwości były wtedy
procesem trwającym wiele godzin, pomimo normalnej perfuzji wieńcowej (okresowy brak
sprzężenia perfuzja-skurcz, perfusion-contraction mismatch). Fakt, że reperfuzja przywracała kurczliwość, dowodził, że komórki sercowe przeżyły stres niedokrwienia i reperfuzji, ale
że uległy przewlekłemu odwracalnemu uszkodzeniu (ogłuszeniu) w postaci przemijających
zaburzeń kurczliwości. Późniejsze badania potwierdziły występowanie ogłuszenia w sercach
wszystkich badanych gatunków zwierząt oraz fakt, że obszary ogłuszone mają prawie prawidłową perfuzję wieńcowa [31-33]. Z badań tych wynika, że:
1. Ogłuszenie jest postacią reperfuzyjnego uszkodzenia mięśnia sercowego. Im poprzedzające niedokrwienie jest większe i trwa dłużej tym kurczliwość w reperfuzji jest mniejsza
i wolniejsza jest jej reperfuzyjna odbudowa. Źródłem ogłuszenia są także łagodne postaci
niedokrwienia towarzyszące anginie wysiłkowej (vide poniżej) [31,36];
2. Ogłuszenia, będące wynikiem powtarzających się incydentów niedokrwienia i reperfuzji,
sumują się w postaci tzw. przewlekłego ogłuszenia [36,37];
3. Czynnikiem toksycznym, wywołującym ogłuszenie są jony Ca2+, które napływają do komórek
sercowych w zwiększonych ilościach w pierwszych paru minutach reperfuzji (rozdz. V.7.). Na
mechanizm ogłuszenia składają dwa następujące Ca2+-zależne mechanizmy (ryc. 5.5.):
a. Trawienie białek kurczliwych, w tym troponiny, przez Ca2+-zależną protezę kalpainę. Troponina w kompleksie z Ca2+ wyzwala skurcz a jej trawienie skutkuje spadkiem jej powinowactwa do Ca2+ i upośledzeniem skurczu. Efekt ten mogą przezwyciężyć interwencje
o działaniu inotropowo dodatnim (np. dobutamina i inne katecholaminy), które zwiększają komórkowe stężenie Ca2+. Podawanie katecholamin w okresie pooperacyjnym jest
powszechnie stosowanym sposobem podtrzymywania funkcji hemodynamicznej serc
operowanych w krążeniu pozaustrojowym. Ustępowanie efektów ogłuszenia jest procesem
długotrwałym gdyż wymaga resyntezy uszkodzonych białek;
b. aktywacja Ca2+-zależnych ATP-az i trwonienie ATP na procesy niezwiązane ze skurczem.
Konsekwencją tego jest zmniejszenie efektywności mechanicznej mięśnia sercowego, co
wobec powolnej reperfuzyjnej odbudowy komórkowych zasobów ATP i CrP, skutkuje osłabieniem skurczu.
Argumenty na rzecz reperfuzyjnego mechanizmu ogłuszenia pochodzą głównie z badań
eksperymentalnych, w których wykazano, że różne interwencje farmakologiczne, zastosowane tuż przed lub tuż po rozpoczęciu reperfuzji, skutecznie zmniejszają ogłuszenie (przyspieszają szybkość powrotu czynności skurczowej w reperfuzji).
86
Ryc. 5.5. Komórkowy mechanizm „ostrego” ogłuszenia. W łagodnej postaci ogłuszenia dominującą
przyczyną zaburzeń kurczliwości może być zmniejszenie efektywności mechanicznej mięśnia. W zaawansowanych postaciach, dominuje enzymatyczne trawienie białek kurczliwych i mniejsza ich wrażliwość na Ca2+. Przyczyną „przewlekłego” ogłuszenia (niepokazane) jest zmniejszona ekspresja białek
kurczliwych.
W patologii ludzkiej większość incydentów niedokrwienia kończy się spontaniczną lub
indukowaną reperfuzją (tab. 5.1.) i wobec tego niedokrwienia te skutkują różnie nasilonym
ogłuszeniem. Klasycznym przykładem ogłuszenia w klinice jest powolny powrót funkcji hemodynamicznej serc poddawanych operacjom kardiochirurgicznym w krążeniu pozaustrojowym [36]. Inny przykład to lokalne odwracalne zaburzenia kurczliwości u osób z CNS
(rozdz. V.5.4)
V.5.4. Lokalne odwracalne zaburzenia kurczliwości u osób z chorobą wieńcową
Wraz z upowszechnieniem się echokardiografii serca, stało się oczywiste, że CNS dość
powszechnie towarzyszą lokalne zaburzenia kurczliwości lewej komory, widoczne w badaniu
echo jako obszary: hipokinezy, akinezy, bądź dyskinezy. W 1974 r., Horn i wsp. [38] pierwsi
wykazali, że część tych słabo kurczących się obszarów odzyskuje kurczliwość pod wpływem
katecholamin. Badanie to obaliło, obowiązujący wtedy dogmat, że przyczyną lokalnych zaburzeń kurczliwości w CNS jest utrata miocytów i zbliznowacenie tkanki. Badanie to wykazało, że, przynajmniej w niektórych słabo kurczących się obszarach, obecne sa ciągle żywe
miocyty, które z jakiegoś powodu aktualnie słabo się kurczą, ale mają rezerwę kurczliwości
wyzwalaną pod wpływem katecholamin. W latach 1985-89 Rahimtoola zaobserwował, że
niektóre słabo kurczące się obszary odzyskują kurczliwość w wyniku poprawy drożności stenotycznej tętnicy wieńcowej (CABG) zaopatrującej te obszary [39]. Równocześnie wysunął
pomysł, że u podstawy „lokalnych odwracalnych zaburzeń kurczliwości” leży częściowe niedokrwienie i stan analogiczny do hibernacji (częściowo niedokrwiony mięsień, aby przeżyć,
wyłącza energochłonny skurcz). Późniejsze badania eksperymentalne potwierdziły istnienie
87
zjawiska „hibernacji” mięśnia sercowego (rozdz. V.5.2.). Niemniej jednak, nowsze badania
nie potwierdziły obecności spoczynkowego niedokrwienia w obszarach serca z lokalnymi
odwracalnymi zaburzeniami kurczliwości. Coraz powszechniejsze jest obecnie przekonanie,
że zaburzenia te są postacią ogłuszenia (a nie hibernacji) wywołanego powtarzającymi się
incydentami niedokrwienia wysiłkowego.
Pierwszy przekonujący dowód na temat roli niedokrwienia wysiłkowego w powstawaniu
ogłuszenia u ludzi pochodzi z pracy Ambrosio i wsp. [40]. W badaniu tym pacjenci z chorobą
wieńcową poddawani byli testowi wysiłkowemu, który u wszystkich badanych powodował
obniżenie odcinka ST elektrokardiogramu, ból wieńcowy oraz lokalne zaburzenia kurczliwości lewej komory (objawy typowe dla niedokrwienia wysiłkowego). Po zaprzestaniu wysiłku
zmiany odcinka ST oraz ból wieńcowy szybko ustępowały. Natomiast zmiany kurczliwości
utrzymywały się jeszcze przez kilkadziesiąt minut, mimo że obszary komory z prawidłową
i nieprawidłową kurczliwością miały taką sama perfuzję wieńcową (scyntygrafia). Takie same
wyniki uzyskano w badaniu z bezpośrednim pomiarem perfuzji wieńcowej przy pomocy pozytronowej emisyjnej tomografii (PET) [41].
Kolejne prace wykazały, że lokalne zaburzenia kurczliwości wywołane przez dwa następujące po sobie incydenty niedokrwienia sprowokowanego wysiłkiem fizycznym [42] lub infuzją dobutaminy [43], sumują się, i że w obszarach „ogłuszonych” i kontrolnych perfuzja jest
taka sama.
W ostatnich latach ukazało się kilkanaście prac, z użyciem PET, badających perfuzję
wieńcową u pacjentów z chorobą wieńcową i lokalnymi odwracalnymi zaburzeniami kurczliwości. PET jest jedyną dostępną obecnie techniką umożliwiającą nieinwazyjny pomiar
perfuzji miokardium w jednostkach bezwzględnych (ml/min/g), co stanowi istotny postęp
w stosunku do technik scyntygraficznych identyfikujących jedynie lokalne gradienty perfuzji.
Pomiary z użyciem PET dość zgodnie pokazują, że obszary miokardium, charakteryzujące się
odwracalnymi zaburzeniami kurczliwości, mają taką samą spoczynkową perfuzję wieńcową
ale zdecydowanie mniejszą rezerwę wieńcową niż obszary kontrolne z prawidłową kurczliwością [44-46]. Przykład takich pomiarów prezentuje Tab. 5.3. W tej i innych pracach, pomiary
dotyczyły słabo kurczących się obszarów z dodatnią próbą dobutaminową, które to obszary
odzyskały kurczliwość w wyniku udrażniającego zabiegu naczyniowego (plastyka wieńcowa
lub CABG).
Tabela 5.3. Spoczynkowy przepływ wieńcowy i rezerwa wieńcowa w kontrolnych obszarach
lewej komory i obszarach z odwracalnymi zaburzeniami kurczliwości u pacjentów z chorobą
wieńcową (wg [44])
Segment
lewej komory
Perfuzja spoczynkowa
(ml/min/g)
Perfuzja po
dipirydamolu
(ml/min/g)
Rezerwa
wieńcowa
(%)
Kontrolny
0,78 ± 0,1
2,38 ± 0,5
305
Ogłuszony
0,88 ± 0,2
1,12 ± 0,4
127
88
Te i inne pomiary pokazują, że obszary z lokalnymi odwracalnymi zaburzeniami kurczliwości mają normalną spoczynkową perfuzję wieńcową, co oznacza, że obserwowane zaburzenia kurczliwości nie są rezultatem spoczynkowego niedokrwienia, ani hibernacji. Aktualna
hipoteza dotycząca mechanizmu tych zaburzeń przyjmuje, że ich przyczyną jest przewlekłe
ogłuszenie, które powstaje w wyniku sumowania się licznych, krótkotrwałych incydentów
niedokrwienia wysiłkowego, jakie towarzyszą aktywności życia codziennego (ryc. 5.6.).
Za słusznością tej hipotezy przemawiają, między innymi, następujące dane eksperymentalne [47]: (i) Podobnie jak u ludzi, także u psów z przewlekłą subkrytyczną stenozą
wieńcową, w obszarze lewej komory za stenozą występują lokalne zaburzenia kurczliwości;
(ii) Obszary słabo kurczące się mają normalną spoczynkową perfuzję wieńcową i ograniczoną
rezerwę wieńcową, co sugeruje, że są to obszary ogłuszone; (iii) Nasilenie lokalnych zaburzeń
kurczliwości ściśle koreluje ze stopniem ograniczenia rezerwy wieńcowej; (iv) Podobnie jak
u ludzi, także u psów, obszary o zmniejszonej kurczliwości reagują dwufazowymi zmianami
kurczliwości na wzrastające dawki dobutaminy.
Ryc. 5.6. Postulowany mechanizm lokalnych odwracalnych zaburzeń kurczliwości lewej komory u osobników z chorobą niedokrwienną serca
Istnieją jedynie ograniczone informacje dotyczące zmian histologicznych w obszarach
serca ludzi z lokalnymi odwracalnymi zaburzeniami kurczliwości (materiał uzyskany przy
pomocy cienkoigłowej biopsji). Początkowo wydawało się, że główną zmianą w tych obszarach jest zanik aparatu kurczliwego kardiomiocytów. Interpretowano tę zmianę jako formę
dostosowywania się kardiomiocytów do stanu bezczynności. W miarę gromadzenia nowych
danych, staje się coraz bardziej oczywiste, że lokalnym zmianom kurczliwości towarzyszą
zmiany histologiczne świadczące o postępującej w czasie degeneracji tkanki [44,45]. Objawia się to, między innymi, postępującym wymieraniem kardiomiocytów i zastępowaniem ich
przez tkankę łączną (tab. 5.4.).
89
Tabela. 5.4. Zmiany morfologiczne w obszarach lewej komory z lokalnymi odwracalnymi
zaburzeniami kurczliwości
• Postępująca utrata aparatu kurczliwego;
• Liczne ziarnistości glikogenu;
• Liczne małe mitochondria obok ziarnistości glikogenu;
• Utrata siateczki śródplazmatycznej;
• Utrata kanalików T;
• Destrukcja szkieletu komórkowego;
• Zmiany w chromatynie jądrowej;
• Apoptoza w subendokardium;
• Postępujące zwłóknienie;
Leczeniem z wyboru lokalnych zaburzeń kurczliwość jest udrożnienie subkrytycznie
zwężonych tętnic wieńcowych (plastyka, CABG). Obserwacje kliniczne pokazują, że powrót
kurczliwości w wyniku takiego zabiegu jest procesem długotrwałym (tygodnie/miesiące)
i często niekompletnym. Przewlekłość procesu wynika prawdopodobnie z faktu, że regeneracja utraconego aparatu kurczliwego (tab. 5.4.) wymaga długiego czasu. Niekompletność
procesu może być konsekwencją postępującego wymierania kardiomiocytów. Oznacza to, że
im wcześniej ma miejsce zabieg naczyniowy tym większa jest jego skuteczność, mierzona
szybkością i kompletnością odbudowy kurczliwości.
V.6. Metabolizm energetyczny serca w niedokrwieniu i reperfuzji
Profil metaboliczny mięśnia sercowego w niedokrwieniu i reperfuzji determinują trzy
czynniki zależne od stopnia ograniczenia perfuzji wieńcowej. Są to:
1. Dostępność tlenu; Tlenowe przemiany są najwydajniejszym sposobem produkcji ATP. Dlatego w mięśniu sercowym, w którym zachowana jest choćby szczątkowa perfuzja wieńcowa, głównym źródłem ATP (choć zwykle niewystarczającym) są ciągle przemiany tlenowe
a nie glikoliza. Eksperymenty zwierzęce wykazały, że dotyczy to także tych obszarów objętych zawałem, w których obecna jest rezydualna perfuzja wieńcowa, związana z krążeniem
obocznym. Glikoliza jest podstawowym źródłem ATP jedynie w obszarach całkowicie pozbawionych perfuzji;
2. Dostępność substratów energetycznych; W obszarach ze znacznym ograniczeniem perfuzji
wieńcowej, dowóz substratów energetycznych z krwią jest ograniczony i wtedy głównym
źródłem glukozy i NEFA stają się wewnątrzkomórkowy glikogen i triglicerydy. Natomiast
obszary niedokrwione z zachowanym niewielkim przepływem a także obszary reperfundowane są eksponowane na zwiększone poziomy NEFA we krwi. Niedokrwienie miokardium, zarówno w przebiegu choroby niedokrwiennej serca, jak i związane z kardiochirurgią i krążeniem pozaustrojowym, skutkuje aktywacją współczulną i często wielogodzinnym
wzrostem poziomu noradrenaliny we krwi. Konsekwencją tego jest zwiększone uwalnianie
NEFA do krwi z tkanki tłuszczowej (poprzez aktywację hormono-zależnej lipazy) (rozdz.
IV.6) oraz zmniejszone uwalnianie insuliny z trzustki i spadek insulinowrażliwości tkanek
[48]. Stan ten może niekorzystnie wpływać na metabolizm mięśnia sercowego. W niedokrwieniu ekspozycja komórek sercowych na zwiększone stężenia NEFA skutkuje, bowiem
90
zwiększoną komórkową akumulacją NEFA i ich toksycznych metabolitów (acylo-CoA
i acetylo-karnityna w większych stężeniach mają toksyczne działanie detergentowe). W reperfuzji, ekspozycja taka skutkuje przedłużającą się komórkową akumulacją kwasu mlekowego i protonów, co utrudnia reperfuzyjną odbudowę czynności mechanicznej serca
(rozdz. V.7.);
3. Akumulacja w miokardium kwasu mlekowego, CO2 i protonów; Im mniejszy jest przepływ
wieńcowy tym mniejsze jest wypłukiwanie tych substancji z niedokrwionych komórek.
Skutkuje to kwasicą komórkową i postępującym zahamowaniem glikolizy mimo równoczesnej obecności różnych aktywatorów glikolizy (rozdz. IV.5.). Inną konsekwencją kwasicy komórkowej jest toksyczna komórkowa akumulacja Ca2+ (rozdz. V.7.).
V.6.1 Metabolizm w niedokrwieniu wysiłkowym
W fizjologii, zwiększone obciążenie mięśnia sercowego (poprzez wysiłek fizyczny, szybką
stymulację, infuzję katecholamin) skutkuje wzrostem przepływu wieńcowego, MVO2, zwiększonym wychwytywaniem i utlenianiem glukozy, kwasu mlekowego oraz kwasów tłuszczowych. Niedokrwieniu wysiłkowemu towarzyszy netto uwalnianie kwasu mlekowego z serca,
co sugeruje aktywację glikolizy beztlenowej [3,4].
Metabolizm energetyczny mięśnia sercowego u ludzi ze stabilną chorobą wieńcową był
badany głównie (przy pomocy metody PET) przed i po wysiłku fizycznym lub przed i po
okresie szybkiej stymulacji serca indukującej niedokrwienie wysiłkowe. Natomiast dane na
temat metabolizmu w trakcie samego niedokrwienia wysiłkowego są bardzo skąpe. Generalnie wydaje się, że w warunkach spoczynkowych metabolizm energetyczny glukozy i kwasów
tłuszczowych nie różni się w zdrowym mięśniu sercowym i w obszarach zaopatrywanych
przez zwężoną tętnicę wieńcową. W czasie wysiłku lub stymulacji serca, w dorzeczu zwężonej
tętnicy obserwowano obszary z deficytem przepływu oraz upośledzonym wychwytywaniem
egzogennej glukozy i utlenianiem egzogennych NEFA. Sugeruje to, że zwiększona aktywność
glikolityczna i uwalnianie kwasu mlekowego towarzyszące niedokrwieniu wysiłkowemu są
wynikiem zwiększonego rozkładu glikogenu. Pomiary wykonane już po zaprzestaniu wysiłku
lub szybkiej stymulacji wykazały zwiększone, w porównaniu z otoczeniem, wychwytywanie
glukozy w dorzeczu zwężonej tętnicy, mimo że przepływ wieńcowy był wszędzie podobny.
Lepiej został zbadany metabolizm niedokrwienia wysiłkowego w warunkach doświadczalnych. Dla przykładu, Stanley i wsp.[49-51] kaniulowli in situ tętnicę przednią zstępującą
(GPZ) u świni i perfundowali dorzecze tętnicy normalnie utlenowaną krwią przy pomocy
pompy. W tak przygotowanym sercu prowokowali niedokrwienie wysiłkowe poprzez równoczesne ograniczenie spoczynkowego przepływu wieńcowego w GPZ o 20% i systemowe
podanie dobutaminy. Innymi słowy, badany obszar miał stałą i ograniczoną o 20% perfuzję wieńcową i dodatkowo był poddany stymulacji katecholaminowej zwiększającej wydatek
energetyczny mięśnia sercowego, co wobec stałości przepływu skutkowało niedokrwieniem
wysiłkowym. W dorzeczu GPZ dotkniętym takim niedokrwieniem:
1. Przepływ wieńcowy i MVO2 pozostawały niezmienione, komórkowe poziomy ATP powoli
malały, a ADP rosły. Równocześnie znacznie malała kurczliwość miokardium i jego efektywność mechaniczna;
2. Wychwytywanie egzogennych NEFA było niezmienione, a ich spalanie zmniejszone;
91
3. Rosły natomiast: wychwytywanie egzogennej glukozy, rozpad glikogenu, netto produkcja
kwasu mlekowego i w niewielkim stopniu utlenianie glukozy. Oznacza to, że w badanym
modelu, niedokrwieniu wysiłkowemu towarzyszyło: (i) przekierowanie metabolizmu energetycznego serca ze spalania NEFA na spalanie glukozy; (ii) aktywacja glikolizy oraz (iii)
częściowy brak sprzężenia glikolizy i utleniania glukozy (spalanie produktów glikolizy nie
nadąża za ich produkcją, o czym świadczy netto produkcja kwasu mlekowego mimo aktywacji spalania glukozy);
4. Częściowe zahamowanie utleniana NEFA przy pomocy inhibitora CPT-I (oxfenicyna)
[49,50,52] lub inhibitora dekarboksylazy malonylo-CoA (który zwiększał komórkowe poziomy malonylo-CoA i w ten sposób hamował aktywność CPT-I [49] skutkowało zwiększonym utlenianiem glukozy, mniejszą produkcją kwasu mlekowego i zwiększoną efektywnością mechaniczną mięśnia sercowego. Sugeruje to, że przyczyną komórkowej akumulacji
kwasu mlekowego i związanego z tym spadku efektywności mechanicznej mięśnia było
hamowanie dehydrogenazy pirogronianowej przez produkty metabolizmu NEFA.
Fakt, że inhibitory utleniania NEFA, takie jak ranolazyna [53] i trimetazydyna [54],
zmniejszają objawy niedokrwienia wysiłkowego u pacjentów ze stabilną chorobą wieńcową
sugeruje, że zaburzenia metabolizmu, podobne do opisanych przez Stanley’a i wsp. u świni,
występują także u człowieka (rozdz. VI.2.4).
V.6.2. Metabolizm w zawale
W mięśniu sercowym dotkniętym niedokrwieniem spoczynkowym i/lub zawałem perfuzja wieńcowa jest ograniczona już w spoczynku. Skutkuje to: (i) deficytem tlenu oraz (ii)
postępującą akumulacją metabolitów w dorzeczu niedrożnej tętnicy i oba te czynniki determinują profil metaboliczny miokardium (ryc. 5.7.).
Konsekwencjami niedoboru tlenu są zwolnienie fosforylacji oksydacyjnej i wtórnie do
tego (i) zmniejszona produkcja i rozkład ATP do AMP i aktywacja kinazy AMP (rośnie komórkowy stosunek stężeń AMP/ATP, rozdz. IV.7) oraz (ii) wzrost komórkowego stosunku
stężeń NADH/NAD+ i FADH2/FAD2+ [22]:
1. Konsekwencje aktywacji AMPK obejmują:
a. aktywację glikolizy i glikolitycznej produkcji ATP w wyniku zwiększonego transportu
błonowego glukozy i zwiększonego rozkładu glikogenu oraz aktywacji glikolizy (poprzez
aktywację heksokinazy i fosfofruktokinazy);
b. aktywację dokomórkowego transportu NEFA oraz zahamowanie karboksylazy acetyloCoA (ACC), co wobec równoczesnego zahamowania β-oksydacji, skutkuje zwiększonym
magazynowaniem NEFA w postaci triglicerydów (ryc. 5.7.A);
c. hamowanie energochłonnych procesów syntezy różnych substancji (rozdz. IV.7.).
2. Wzrost stosunku NADH/NAD+ i FADH2/FAD2+ skutkuje:
a. zwolnieniem cyklu Krebsa, β-oksydacji NEFA i utleniania pirogronianu (na poziomie
PDH) oraz
b. aktywacją dehydrogenazy mleczanowej i zwiększoną produkcją kwasu mlekowego z pirogronianu. Aktywna dehydrogenaza mleczanowa zapewnia regenerację NAD+ z NADH,
co umożliwia nieprzerwane funkcjonowanie enzymów glikolitycznych i beztlenową produkcję ATP. Początkowo glikolityczna produkcja ATP rzeczywiście rośnie. Następnie,
92
w miarę gromadzenia się kwasu mlekowego i protonów w niedokrwionym obszarze,
znowu maleje (vide poniżej).
W ciągu 30 sekund po podwiązaniu tętnicy wieńcowej u psa, glikoliza i produkcja kwasu
mlekowego w niedokrwionym obszarze rosną nawet piętnastokrotnie. Odpowiadają za to glikogenoliza, aktywacja enzymów glikolitycznych, a obszarach z rezydualną perfuzją wieńcową, dodatkowo jeszcze zwiększony dokomórkowy transport glukozy. Z czasem, szybkość glikolizy maleje i w 30. minucie jest już tylko nieznacznie zwiększona, a w 60. minucie jest nawet
mniejsza niż w normalnym obszarze. Jest to spowodowane wyczerpywaniem się zapasów glikogenu, a częściowo hamowaniem aktywności fosfofruktokinazy i dehydrogenazy aldehydu
3-fosfoglicerynowego, dwóch kluczowych enzymów glikolitycznych, przez gromadzący się
w komórkach kwas mlekowy, jony H+ i NADH. Wzrost komórkowego stężenia H+ (kwasica
komórkowa) hamuje fosfofruktokinazę a komórkowa akumulacja mleczanów i NADH skutkuje hamowaniem dehydrogenazy aldehydu 3-fosfoglicerynowego [55].
Akumulacja tych inhibitorów glikolizy rośnie proporcjonalnie do stopnia ograniczenia
perfuzji wieńcowej i ich utrudnionego wypłukiwania z niedokrwionej tkanki.
Ostatecznie, w ognisku niedokrwienia tlenowa produkcja ATP drastycznie maleje, a glikolityczna rośnie. Niemniej jednak, produkcja tlenowa jest ciągle dominującym, choć niewystarczającym, źródłem ATP w obszarach z rezydualną perfuzją wieńcową. Jedynie w obszarach bez perfuzji, glikoliza staje się dominującym i jedynym źródłem ATP. Niezależnie od
wielkości niedokrwienia, produkcja ATP w ognisku niedokrwienia jest niewystarczająca do
utrzymania prawidłowego poziomu kurczliwości miokardium.
V.6.3. Metabolizm w reperfuzji
Cztery elementy decydują o charakterystycznym profilu metabolicznym reperfundowanego mięśnia sercowego [22]. Są to:
1. Reoksygenacja niedokrwionego obszaru, która sprawia, że:
a. mitochondria wznawiają fosforylację oksydacyjną i produkcję ATP. Wymaga to dostępności
tlenu i substratów do produkcji ATP, jakimi są ADP i AMP. Komórkowa pula ADP i AMP
jest jednak zmniejszona w związku z ich reperfuzyjnym wypłukiwaniem z mięśnia (rozdz.
V.3.). Wykazano, że reperfuzja serc operowanych w krążeniu pozaustrojowym płynem zawierającym prekursory syntezy AMP, przyspieszała odbudowę funkcji serc w reperfuzji;
b. mitochondria wznawiając fosforylację oksydacyjną zmniejszają komórkowe poziomy
NADH i FADH2, co skutkuje reaktywacją cyklu Krebsa i β-oksydacji. Spadek poziomu
NADH powinien skutkować także reaktywacją dehydrogenazy pirogronianowej (PDH)
i spalania glukozy. PDH pozostaje jednak zahamowana przez produkty reaktywowanej
β-oksydacji;
c. mitochondria wznawiając fosforylacjię oksydacyjną w sytuacji kiedy komórkowe poziomy NADH i FADH2 są duże a poziom ADP niski, stają się producentem dużej ilości
toksycznych ROS, które wespół z innymi czynnikami, aktywują megakanał mitochondrialny, z wszystkimi niekorzystnymi tego konsekwencjami dla reperfundowanego miokardium (rozdz. V.8);
2. Wypłukiwanie z niedokrwionego obszaru inhibitorów glikolizy (kwas mlekowy, protony),
co stwarza warunki do reaktywacji glikolizy;
93
A.
B
Ryc. 5.7. Metabolizm energetyczny miokardium w niedokrwieniu spoczynkowym i zawale oraz w reperfuzji. (A) W obszarze z rezydualną perfuzją: (i) fosforylacja oksydacyjna jest zwolniona, ale pozostaje
dominującym źródłem ATP; (ii) dokomórkowy transport NEFA rośnie, ale spalanie NEFA maleje i komórkowe poziomy acylo-CoA i acylo-karnityny rosną, co faworyzuje syntezę triglicerydów oraz skutkuje toksycznym detergentowym działaniem tych substancji na błony komórkowe; (iii) Glikoliza jest przyspieszona, ale spalanie glukozy jest zwolnione na poziomie PDH (wtórnie do komórkowej akumulacji
NADH); (iv) Ten stan rozprzęgania procesów glikolizy i spalania glukozy skutkuje akumulacją kwasu
mlekowego i protonów i z czasem wtórnym hamowaniem glikolizy. (B) W reperfuzji następuje reaktywacja (przynajmniej częściowa) fosforylacji oksydacyjnej i β-oksydacji NEFA. Produkty β-oksydacji
hamują PDH, co sprawia, że proces rozprzęgania glikolizy i spalania glukozy i zwiększonej produkcji
kwasu mlekowego, paradoksalnie, dotyczy także okresu reperfuzji.
94
3. Wysoka aktywność AMPK, częściowo odziedziczona po okresie niedokrwienia. Ostatnio
okazało się, że ROS aktywują AMPK w sposób niezależny od AMP [56], co może być dodatkowym mechanizmem wzrostu aktywności AMPK w reperfundowanym miokardium.
Konsekwencjami stymulacji AMPK w reperfuzji są: duża aktywność glikolizy β-oksydacji
i równoczesne zahamowanie PDH przez produkty β-oksydacji (ryc. 5.7.B). W efekcie, mimo
reaktywacji metabolizmu tlenowego, proces rozprzęgania glikolizy i spalania glukozy i zwiększonej produkcji kwasu mlekowego dotyczy także okresu reperfuzji (PDH nie nadąża z metabolizmem zwiększonych ilości pirogronianu, który jest przekształcany w kwas mlekowy).
Aktywacja β-oksydacji w reperfuzji jest konsekwencją zahamowania przez AMPK karboksylazy acetylo-CoA (ACC), mniejszej produkcji malonylo-CoA i odhamowania CPT I (enzym
odpowiedzialny za transport NEFA do mitochondriów (rozdz. IV.4.);
4. Komórkowa akumulacja kwasu mlekowego (skutkująca kwasicą komórkową i jej toksycznymi
konsekwencjami, rozdz. V.7.), wtórna do rozprzęgania procesów glikolizy i spalania glukozy.
V.7. Komórkowa kwasica i toksyczna akumulacja Ca2+ w niedokrwieniu
i reperfuzji
1. Kwasica komórkowa towarzysząca niedokrwieniu i reperfuzji bezpośrednio upośledza kurczliwość
mięśnia sercowego a pośrednio prowadzi do szkodliwej komórkowej akumulacji Ca2+;
2. W rezultacie tzw. paradoksu pH, akumulacja Ca2+ jest szczególnie intensywna i toksyczna
w pierwszych paru minutach reperfuzji, kiedy staje się ważną przyczyną reperfuzyjnego
uszkodzenia serca;
3. Na mechanizm toksyczności Ca2+ składają się:
(a) „trwonienie” ATP na procesy niezwiązane ze skurczem, co upośledza efektywność mechaniczną mięśnia i opóźnia odbudowę jego funkcji w reperfuzji, mimo powrotu prawidłowej
perfuzji wieńcowe (element mechanizmu ogłuszenia);
(b) aktywacja Ca2+-zależnych proteaz i fosfolipaz, skutkująca destrukcją białek i/lub błon komórkowych (element mechanizmu ogłuszenia);
(c) aktywacja aparatu skurczowego skutkująca nadmiernym skurczem kardiomiocytów
uszkadzającym błony komórkowe i umożliwiającym niekontrolowany dokomórkowy napływ Ca2+ i tzw. martwica z węzłami skurczu (contraction-band necrosis);
(d) Ca2+-zależna aktywacja megakanału mitochondrialnego skutkująca rozprzęganiem fosforylacji oksydacyjnej i apoptozą.
V.7.1. Homeostaza jonów Ca2+ i H+ w niedokrwieniu i reperfuzji; Paradoks pH
W czasie każdego pobudzenia otwierają się błonowe kanały wapniowe i do kardiomiocytów napływają jony Ca2+, które wraz z Ca2+ uwalnianym z siateczki śródplazmatycznej, aktywują skurcz. W czasie diastole Ca2+ jest usuwany z komórki przy pomocy błonowego wymiennika Na+/Ca2+. Jego czynność polega na tym, że do komórki napływają trzy jony Na+ (zgodnie
z przezbłonowym gradientem elektrochemicznym Na+) w zamian za jeden jon Ca2+ usuwany
z komórki wbrew gradientowi elektrochemicznemu Ca2+. Źródłem energii dla tego transportu
jest gradient elektrochemiczny Na+ faworyzujący napływ Na+ do komórki i warunkujący efektywność odkomórkowego transportu Ca2+. W podobny sposób, ale przy udziale błonowego wy95
Ryc. 5.8. Homeostaza jonów Na+, Ca2+ i H+ w normie, niedokrwieniu i reperfuzji. Szczegóły w tekście.
96
miennika Na+/H+, z komórki usuwane są protony na wymianę z Na+. Jony Na+ są następnie usuwane z komórki przez pompę sodowo-potasową. Ostatecznie, właśnie pompa Na+/K+ zapewnia
stałość komórkowych stężeń jonów Na+ i K+, a pośrednio, także jonów Ca2+ i H+, (ryc. 5.8.).
W niedokrwieniu dochodzi do zwiększonej produkcji kwasu mlekowego i aktywacji wymiennika Na+/H+ przez gromadzące się w komórce protony oraz do równoczesnego upośledzenia czynności pompy Na+/K+ (z powodu niedoboru ATP). W efekcie, transportowi H+
poza komórkę towarzyszą: (i) postępujący wzrost komórkowego stężenia Na+ (rośnie naływ
Na+ a pompa Na+/K+ jest mniej aktywna); (ii) zmniejszenie przezbłonowego gradientu stężeń
Na+ (iii) wtórne do mniejszego gradientu Na+, zwolnienie czynności wymiennika Na+/Ca2+
i ostatecznie (iv) postępujące gromadzenia się w komórce jonów Ca2+.
Komórkową akumulację Ca2+ w niedokrwieniu częściowo ogranicza sama kwasica komórkowa, która jest czynnikiem hamującym błonowe kanały wapniowe. Ostatecznie, wraz
z przedłużaniem się niedokrwienia narasta wewnątrz- i zewnątrzkomórkowa kwasica (bo
wypłukiwanie H+ z przestrzeni pozakomórkowej jest upośledzone w niedokrwieniu), wewnątrzkomórkowe stężenie Na+ i w umiarkowanym stopniu, wewnątrzkomórkowe stężenie
Ca2+ (ryc. 5.8. Niedokrwienie).
Mechanizmy jonowe typowe dla niedokrwienia, intensyfikują się w czasie reperfuzji,
głównie, dlatego że reperfuzja skutkuje szybszym wypłukiwaniem H+ (i normalizacją pH)
z przestrzeni zewnątrzkomórkowej niż wewnątrzkomórkowej niedokrwionego obszaru. Powoduje to kolejno: (i) gwałtowny wzrost przezbłonowego gradientu stężeń H+, który jest silnym aktywatorem wymiennika Na+/H+; (ii) szybki dokomórkowy napływ jonów Na+, które
nie są wystarczająco szybko usuwane z komórki przez ciągle mało aktywną pompę Na+/K+ oraz
(iii) wtórne do komórkowej akumulacji Na+, odwrócenie kierunku transportu przez wymiennik Na+/Ca2+ i gwałtowny „toksyczny” wzrost komórkowego stężenia jonów Ca2+ w pierwszych paru minutach reperfuzji. Dobrym potwierdzeniem przedstawionego mechanizmu są
eksperymenty pokazujące, że reperfuzja kwaśnym płynem oraz zahamowanie wymiennika
Na+/H+ lub Na+/Ca2+ przeciwdziałają obładowaniu komórek sercowych Ca2+ i reperfuzyjnemu uszkodzeniu serca [57-59].
Przedstawiony mechanizm znany jest jako reperfuzyjny paradoks pH. Polega on na tym, że
reperfuzja, która ostatecznie likwiduje szkodliwą dla komórek kwasicę, paradoksalnie szkodzi
komórkom poprzez ich (i) alkalizację, która umożliwia aktywację różnych potencjalnie szkodliwych procesów hamowanych dotąd przez niskie pH (np. aktywacja kalpaliny, megakanału
mitochondrialnego, apoptozy) oraz (ii) obładowywanie komórek jonami Ca2+. Badania eksperymentalne wykazały, że o losach reperfundowanych komórek decyduje tempo normalizacji
komórkowego pH i Ca2+. Szybsza normalizacja pH niż Ca2+ przeważa szalę na rzecz śmierci komórek. Natomiast szybsza normalizacja Ca2+ niż pH jest dobrym prognostykiem reperfuzyjnej
odbudowy czynności komórek. Czynnikiem decydującym o tempie normalizacji komórkowego
Ca2+ jest tempo reaktywacji pompy Na+/K+, które zależy głównie od dostępności ATP i poziomu
degradacji jej białka przez Ca2+-zależną kalpainę (rozdz. V.7.2) [60,61].
V.7.2. Toksyczne efekty komórkowej akumulacji Ca2+
Komórkowa akumulacja Ca2+ towarzysząca niedokrwieniu, a zwłaszcza reperfuzji, ma
liczne niekorzystne konsekwencje (ryc. 5.9.) [60,61], z których najważniejsze to:
97
Ryc. 5.9. Procesy aktywowane przez toksyczną akumulację Ca2+ w reperfundowanych komórkach
i zmiany czynnościowe z tym związane. Szczegóły w tekście.
1. Aktywacja licznych ATP-az aktywowanych przez Ca2+; Chodzi tu zwłaszcza o aktywację
energochłonnej ATP-az wapniowej siateczki śródplazmatycznej odpowiedzialnej za usuwanie nadmiaru Ca2+ z cyplazmy. W ten sposób komórkowe zasoby ATP zostają w części „przekierowane” z pracy hemodynamicznej pompy sercowej na pracę między innymi
pomp jonowych. Ten efekt „trwonienia” ATP przyczynia się do spadku sprawności mechanicznej mięśnia sercowego. Objawia się to większym spadkiem kurczliwości w niedokrwieniu i/lub wolniejszą i mniej kompletną odbudową czynności skurczowej mięśnia sercowego
w reperfuzji. W bardziej zaawansowanych postaciach komórkowej akumulacji Ca2+, może
dochodzić do nadmiernej aktywacji aparatu kurczliwego kardiomiocytów i przetrwałego
skurczu (tzw. przykurcz, widziany jako zmniejszenie podatności rozkurczowej miokardium). Przykurcz ten może być tak masywny, że powoduje rozrywanie błony komórkowej
kardiomiocytów i „martwicę z węzłami skurczu” (contraction band necrosis).
2. Aktywacja komórkowych Ca2+-zależnych proteaz [60,61]; Dobrze udokumentowany jest
fakt aktywacji w niedokrwionym/reperfundowanym miokardium Ca2+-zależnej kalpainy,
enzymu proteolitycznego trawiącego między innymi białka aparatu kurczliwego komórek
(w tym troponinę I) oraz białka błony komórkowej (w tym ATP-azę sodowo-potasową).
Trawienie troponiny jest elementem mechanizmu ogłuszenia mięśnia sercowego (spadek
wrażliwości aparatu kurczliwego na Ca2+ i przejściowy spadek kurczliwości miokardium towarzyszący reperfuzji) (rozdz. III.5.3) [62,63]. Trawienie ATP-azy opóźnia transport jonów
i przyczynia się do zwiększonej i przedłużającej się komórkowej akumulacji Ca2+. Trawienie
białek błonowych z jednej strony zwiększa podatność błon komórkowych na rozrywanie,
czym przyczynia się do łatwiejszego ich pękania i śmierci komórek w reperfuzji. Z drugiej
strony wykazano, że kalpaina trawi białka anty-apoptotyczne (Bid) i równocześnie akty98
wuje białka promujące apoptozę (Bad i Bax). Czynnikiem aktywującym kalpainę w czasie
reperfuzji, obok Ca2+, jest także alkalizacja komórek w reperfuzji (niskie pH jest inhibitorem kalpainy). W tym kontekście wykazano, że przedłużająca się kwasica w reperfuzji (np.
poprzez kwaśną reperfuzję), zapobiega reperfuzyjnej aktywacji kalpainy, proteolizie w reperfuzji i reperfuzyjnemu uszkodzeniu miokardium. Oznacza to, że aktywacja kalpainy ma
miejsce dopiero w reperfuzji. Fakt ten otwiera możliwości blokowania enzymu tuż przed
zabiegiem reperfuzji. Pierwsze zachęcające próby takiego blokowania przeprowadzono już
w eksperymentach zwierzęcych [60,61].
Ryc. 5.10. Wzory strukturalne glicerolu i jego pochodnych: triacylogliceroli i fosfolipidów. R1-3 – cząsteczki kwasów tłuszczowych; czarne prostokąty – wymieniają nazwy czterech typów fosfolipaz rozkładających fosfolipidy; X – zasada nieorganiczna (kilka ich rodzajów buduje fosfolipidy i one decydują
o specyficznych właściwościach danego fosfolipidu). Aktywacja Ca2+ – zależnej fosfolipazy A2 skutkuje
powstaniem toksycznego lizofosfatydu i wolnego wielonienasyconego kwasu tłuszczowego, najczęściej
kwasu arachidonowego.
3. Aktywacja Ca2+-zależnych fosfolipaz; Dobrze udokumentowany jest fakt aktywacji, pod
wpływem nawet krótkotrwałych incydentów niedokrwienia, Ca2+-zależnej fosfolipazy A2.
Enzym ten odłącza od cząsteczki fosfolipidu resztę kwasu tłuszczowego związaną ze środkowym węglem glicerolu(ryc. 5.10.). Powstają wtedy: toksyczny związek lizofosfatyd [64],
oraz NEFA, którym z reguły jest wielonienasycony kwas tłuszczowy, np. kwas arachidonowy. Trawienie fosfolipidów osłabia strukturę błon komórkowych i zwiększa ich podatność
na rozrywanie. Strukturę błon osłabiają także lizofosfatydy, które są substancjami o silnych
właściwościach detergentowych. Aktywacja fosfolipazy A2, poprzez uwalnianie kwasu arachidonowego, uruchamia tzw. kaskadę kwasu arachidonowego, której produktami są prostaglandyny i trombksany. Te ostatnie są substancjami obkurczającymi naczynia i aktywującymi płytki, przez co mogą pogłębiać aktualnie istniejące niedokrwienie lub utrudniać
powrót perfuzji miokardium w reperfuzji (vide no-reflow phenomenon, rozdz. V.10).
4. Aktywacja systemów komórkowych odpowiedzialnych za produkcję toksycznych ROS;
ROS upośledzają czynność pompy Na+/K+, ATP-azy wapniowej w siateczce śródplazma99
tycznej, wymiennika Na+/ Ca2+ oraz białek kurczliwych [65]. Konsekwencją tych zmian
może być dalsze zwiększanie się komórkowego poziomu Ca2+ i powstawanie błędnego koła
regulacyjnego, w którym zwiększona komórkowa akumulacja Ca2+ sprzyja jeszcze większej
akumulacji Ca2+.
5. Ca2+-zależna aktywacja megakanału mitochondrialnego, która skutkuje rozprzęganiem
fosforylacji oksydacyjnej i zaburzeniami mitochondrialnej produkcji ATP oraz aktywacją apoptozy. Jony Ca2+ i stres oksydacyjny są aktywatorami a niskie pH jest inhibitorem
megakanału. Reperfuzji towarzyszy równoczesny wzrost komórkowego pH i Ca2+ i oba te
czynniki biorą udział w reperfuzyjnej aktywacji megakanału (vide podobny mechanizm
aktywacji kalpainy). Podtrzymywanie kwasicy w reperfuzji, np. poprzez kwaśną reperfuzję, zapobiega reperfuzyjnej aktywacji megakanału i reperfuzyjnemu uszkodzeniu miokardium, co dowodzi, że aktywacja megakanału następuje dopiero w reperfuzji. Podobnie, tzw.
hartowanie reperfuzją (ischemic postconditioning) opóźnia normalizację komórkowego
pH w reperfuzji i w ten sposób zapobieganiu reperfuzyjnej aktywacji megakanału [60,61]
(rozdz. V.8).
V.8. Mitochondria źródłem ATP, ale również ROS, apoptozy
i reperfuzyjnego uszkodzenia
Mitochondria stanowią ~30% objętości kardiomiocytów i są odpowiedzialne za >95% sercowej
produkcji ATP niezbędnej dla prawidłowego funkcjonowania pompy sercowej. Obok kurczliwości,
także ostateczne losy niedokrwionego/reperfundowanego kardiomiocyta zależą od zachowania
czynności jego mitochondriów. Jest to konsekwencja faktu, że obok produkcji ATP, mitochondria
mają 3 inne funkcje ważne z punktu widzenia (pato)fizjologii serca. Są to:
1. Produkcja wolnych rodników tlenowych; Fosforylacja oksydacyjna, która jest procesem odpowiedzialnym za produkcję ATP jest równocześnie głównym źródłem anionorodnika ponadtlenkowego (O2–) w kardiomiocytach, przekształcanego następnie w nadtlenek wodoru. Te
tzw. reaktywne formy tlenu (ROS), produkowane przez mitochondria w niewielkich ilościach,
pełnią rolę przekaźników informacji w różnych szlakach sygnalizacji komórkowej. W stanach
patologicznych (niedokrwienie reperfuzja, hiperglikemia, starzenie) produkcja ROS wzrasta
i stają się one toksyczne. Dlatego komórki bogate w mitochondria dysponują rozbudowanym
systemem ochrony anty-rodnikowej;
2. Wyzwalanie procesu apoptozy oraz integracja i wzmacnianie poza mitochondrialnych sygnałów pro-apoptotycznych;
3. Rola końcowego efektora reperfuzyjnego uszkodzenia miokardium; W warunkach wzrostu
stężenia Ca2+ w cytoplazmie, jaki towarzyszy np. aktywności fizycznej czy aktywacji współczulnej, Ca2+ przechodzi do macierzy mitochondrialnej gdzie: (i) jest magazynowany (mitochondria jako komórkowy bufor Ca2+); (ii) zwiększa aktywność metaboliczną mitochondriów
poprzez aktywację kluczowych enzymów regulatorowych i w ten sposób zapewnia dostosowywanie się produkcji ATP do jego rozkładu oraz (iii) w warunkach znacznego obładowania mitochondriów Ca2+, powoduje otwarcie megakanałów mitochondrialnych, co skutkuje
utratą zdolności mitochondriów do produkcji ATP oraz wypływem z mitochondriów białek
inicjujących apoptozę. Aktywacja megakanału, przez Ca2+ i inne czynniki towarzyszące re100
perfuzji, jest kluczowym elementem mechanizmu reperfuzyjnego uszkodzenia miokardium.
Interwencje zapobiegające reperfuzyjnej aktywacji megakanału redukują wielkość martwicy
zawałowej o 30–50%. Różne formy hartowania miokardium działają poprzez zapobieganie
reperfuzyjnej aktywacji megakanału.
V.8.1. Fosforylacja oksydacyjna i produkcja ATP
Fosforylacja oksydacyjna jest procesem, w którym zespół enzymów oksydoredukcyjnych, związanych z wewnętrzną błoną mitochondrialną i tworzących tzw. łańcuch oddechowy (składający się z kompleksów mitochondrialnych I, II, III i IV) (ryc. 5.11.):
Ryc. 5.11. Schemat tlenowych przemian substratów energetycznych i fosforylacji oksydacyjnej. Produktem spalania NEFA, glukozy i kwasu mlekowego są zredukowane kofaktory NADH i FADH2. Łańcuch
oddechowy związany z wewnętrzną błoną mitochondrialną przenosi z NADH i FADH2: (i) elektrony na
O2 (via kompleksy I–IV) oraz (ii) protony do przestrzeni międzybłonowej (kompleksy I, III i IV), co buduje gradient elektrochemiczny H+ w poprzek wewnętrznej błony (ΔΨm). H+ wracają do macierzy przy
udziale kompleksu syntazy ATP zgodnie z ΔΨm, czemu towarzyszy fosforylacja ADP do ATP. Aktywacja mitochondrialnych białek rozprzęgających (UCP) i/lub megakanału „rozładowuje” ΔΨm, co skutkuje
konsumpcją O2 bez produkcji ATP, czyli stanem rozprzęgania fosforylacji oksydacyjnej.
1. transportuje elektrony z NADH i FADH2 poprzez kolejne kompleksy mitochondrialne I–IV
i w kompleksie IV (oksydaza cytochromowa) dochodzi do tzw. czteroelektronowej redukcji
O2 i powstania wody. Niewielka ilość elektronów „wycieka” z łańcucha oddechowego na
poziomie kompleksów I, II i III i reagując z O2 prowadzi do powstania anionorodnika ponadtlenkowego, O2– (rozdz. V.8.2);
2. transportuje protony z NADH i FADH2 z macierzy mitochondrialnej do przestrzeni międzybłonowej, co sprawia, że wnętrze mitochondriów ma potencjał ujemny w stosunku do
potencjału komórkowego, że stężenie protonów w macierzy jest mniejsze niż w przestrzeni
międzybłonowej, i że wobec tego istnieje gradient elektrochemiczny H+ (ΔΨm) faworyzujący przechodzenie H+ z powrotem do macierzy mitochondrialnej;
101
3. transportuje protony, przy udziale kompleksu enzymatycznego syntazy ATP i zgodnie z ich
ΔΨm, z przestrzeni międzybłonowej z powrotem do macierzy, czemu towarzyszy fosforylacja
ADP do ATP (protonowy gradient elektrochemiczny jest źródłem energii dla syntezy ATP).
Ważną cechą systemu produkującego ATP jest to, że przy braku ΔΨm lub, kiedy jest on niewielki, reakcja katalizowana przez syntazę ATP odwraca się i zamiast produkować, enzym
rozkłada ATP do ADP i Pi [66]. W niedokrwionym miokardium wykazano, że syntaza ATP
pracująca w odwróconym trybie konsumuje ATP powstający poza mitochondriami w procesie glikolizy [67], co dodatkowo zmniejsza efektywność mechaniczną mięśnia sercowego.
Istnieje ścisłe sprzężenie transportu elektronów z NADH i FADH2 na O2 w łańcuchu oddechowym, transportu H+ do przestrzeni międzybłonowej oraz procesu fosforylacji ADP do
ATP. Dlatego o wielkości mitochondrialnej produkcji ATP decydują: (i) wielkość ΔΨm, który
jest pochodną dostępności NADH i FADH2 oraz O2 oraz (ii) poziomy ADP i fosforu nieorganicznego (Pi) w macierzy mitochondrialnej, które zależą od wielkości zużycia ATP w aparacie
kurczliwym i szybkości transportu ADP do macierzy przez translokazę ATP/ADP (ryc. 4.2.B).
Produkcja ATP zwalnia, kiedy maleje dostępność NADH i FADH2, O2 i/lub ADP i Pi.
Wielkość ΔΨm kontrolują różne białka wewnętrznej błony mitochondrialnej tworzące
w niej kanały jonowe lub pory. Szczególnym zainteresowaniem cieszą się obecnie megakanał
mitochondrialny (mPTP, mitochondrial permeability transition pore) i tzw. białka rozprzęgające (ang. UCPs, uncoupling proteins). Tworzone przez nie kanały/pory umożliwiają napływ
H+ do macierzy, co skutkuje zmniejszeniem ΔΨm i produkcji ATP, mimo że mitochondria
ciągle konsumują O2. Jest to stan określany jako rozprzęganie fosforylacji oksydacyjnej, któremu na poziomie całego serca towarzyszy zmniejszenie efektywności mechanicznej mięśnia
sercowego (normalna konsumpcja O2 i zmniejszona produkcja ATP).
W sercu obecne są izoformy UPC2 i UPC3. Wiadomo o nich, że są wybiórczymi nośnikami protonów, że są hamowane przez ATP i ADP i aktywowane przez ROS i NEFA (ROS
i NEFA rozprzęgają mitochondria i zmniejszają efektywność mechaniczną serca). Ale generalnie ich rola w fizjologii serca nie jest wyjaśniona. Istnieje natomiast coraz więcej danych
o udziale megakanału mitochondrialnego w mechanizmie apoptozy (rozdz. V.8.3) oraz reperfuzyjnego uszkodzenia miokardium (rozdz. V.8.4).
V.8.2. Mitochondria, wolne rodniki tlenowe i komórkowa ochrona anty-rodnikowa
W normie ~99% O2 konsumowanego przez mitochondria ulega kompletnej czteroelektronowej redukcji do H2O przy udziale oksydazy cytochromowej (kompleks IV) (ryc. 5.12.).
Pozostały 1% O2 ulega niekompletnej, jednoelektronowej redukcji spowodowanej przenoszeniem elektronów na O2 już na wcześniejszych etapach łańcucha oddechowego (kompleksy I,
II i III). Powstaje wtedy cząsteczka zawierająca niesparowany elektron, znana jako anionorodnik ponadtlenkowy (O2–). Atomy, cząsteczki i związki chemiczne zawierające niesparowany
elektron określane są jako wolne rodniki. Są to substancje o dużej reaktywności chemicznej
(łatwo oddają niesparowany elektron na inne cząsteczki) i dlatego cechują się dużą reaktywnością chemiczną, małą trwałością i cytotoksycznością. Powstanie O2– zapoczątkowuje kaskadę reakcji wolnorodnikowych, której konsekwencją jest spontaniczne, lub wspomagane
enzymatycznie, powstanie kolejnych ROS, w tym nadtlenku wodoru oraz rodnika wodorotlenowego (ryc. 5.12.).
102
Ryc. 5.12. Kompletna i jednoelektronowa redukcja O2 towarzyszące fosforylacji oksydacyjnej. Przyłączenie elektronu do O2 skutkuje powstaniem anionorodnika ponadtlenkowego (O2–), co inicjuje kaskadę reakcji wolnorodnukowych i powstają kolejno: nadtlenek wodoru (H2O2), rodnik wodorotlenowy
(.OH) i H2O. W nadmiarze, ROS (O2–, H2O2 i .OH) są toksyczne. Strażnikami ich stężenia są enzymy
dyzmutaza ponadtlenkowa (SOD, przekształca O2– w H2O2) oraz katalaza i peroksydaza glutationu,
przekształcające H2O2 do H2O, bez udziału .OH.
Wiadomo obecnie, że ROS pochodzenia mitochondrialnego pełnią role przekaźników
informacji w różnych (pato)fizjologicznych szlakach sygnalizacji komórkowej. Dla przykładu, biorą udział w mechanizmie hartowania niedokrwieniem (rozdz. VI.3) i apoptozy (rozdz.
V.8.3). Mitochondria są także głównym źródłem nadprodukcji ROS i stresu oksydacyjnego
w niedokrwionym/reperfundowanym miokardium. Jedną z konsekwencji tego stresu jest aktywacja megakanału mitochondrialnego (rozdz. V.8.4).
Dokładny molekularny mechanizm mitochondrialnej produkcji O2– jest słabo poznany.
Regułą jest jednak, że im większe jest komórkowe zapotrzebowanie na ATP (czego miarą
jest wysoki mitochondrialny poziom ADP i Pi), tym szybszy jest przepływ elektronów przez
łańcuch oddechowy i trudniejsze są warunki do wyciekania elektronów z łańcucha oddechowego i tworzenia O2–. Można wymienić przynajmniej dwie sytuacje (pato)fizjologiczne,
którym towarzyszy zwolniony przepływ elektronów w łańcuchu oddechowym i zwiększona
produkcja O2–. Są to:
(i) Niedokrwienie i reperfuzja. Wraz z niedoborem tlenu rośnie komórkowy poziom zredukowanych kofaktorów NADH i FADH2 (NADH i FADH2 > O2) i poziom redukcji różnych
komponent łańcucha oddechowego. Sprzyja to wyciekowi elektronów i tworzeniu O2– już
w okresie niedokrwienia (z udziałem resztkowego O2), a następnie dużych ilości O2– w początkowym okresie reperfuzji, kiedy dostępność O2 jest już znowu duża (stan znany jako
reperfuzyjny paradoks tlenowy);
(ii) Stany, w których rozkład ATP do ADP i Pi jest wolniejszy niż mitochondrialna produkcja
NADH i FADH2 (NADH i FADH2 > ADP i Pi). W warunkach fizjologicznych, nierówność
taka może być konsekwencją nadmiernej ekspozycji komórek na substraty energetyczne
(hiperglikemia, podwyższony poziom NEFA w surowicy). Inny powód to zbyt mała aktywność fizyczna i rozkład ATP do ADP i Pi w stosunku do normalnej produkcji NADH
i FADH2. W konsekwencji, rosną wtedy ΔΨm i mitochondrialna akumulacja ATP. W tym
103
kontekście wykazano, że łagodne rozprzęganie fosforylacji oksydacyjnej (np. poprzez aktywację UCP przez ROS czy NEFA), choć zmniejsza produkcję ATP, redukuje mitochondrialną produkcję O2–, co jest prawdopodobnie mechanizmem ochronnym. Nierówność
NADH i FADH2 > ADP i Pi jest typowa również dla reperfundowanego miokardium,
w którym poziomy ADP i Pi maleją najpierw ze względu na niedokrwienną degradację
ATP, a następnie z powodu zwiększonego wypłukiwaniu jego metabolitów z miokardium
w reperfuzji (rozdz. V.3.).
Komórki czerpiące energię z przemian tlenowych, w tym zwłaszcza kardiomiocyty, są
wyposażone w system ochrony antyrodnikowej, na który składają się: (i) enzymy neutralizujące ROS (ryc. 5.12.); (ii) drobnocząsteczkowe substancje redukujące, które usuwają ROS
w sposób nieenzymatyczny (tzw. wymiatacze wolnych rodników, jak kwas askorbinowy,
α-tokoferol, β-karoten, zredukowany glutation) oraz prawdopodobnie (iii) mitochondrialne
systemy rozprzęgające fosforylację oksydacyjną (UPC, mega kanał mitochondrialny, kanały
jonowe w wewnętrznej błonie mitochondrialnej) (rozdz. VIII).
V.8.3. Mitochondria vs. apoptoza
Generalnie apoptoza jest procesem fizjologicznym, podczas którego z organizmu eliminowane są nadliczbowe, starzejące się, uszkodzone lub zmienione nowotworowo komórki. Okazuje się, że zwiększone wymieranie kardiomiocytów w mechanizmie apoptozy jest
elementem patomechanizmu także różnych chorób układu sercowo-naczyniowego. Nasilenie apoptozy wykazano w miokardium pacjentów z zawałem, kardiomiopatią cukrzycową
i krańcową postacią zastoinowej niewdolności serca. Badania eksperymentalne sugerują, że
apoptoza dotyczy głównie kardiomiocytów w strefie granicznej między zawałem i prawidłowo ukrwionym miokardium (border zone) oraz, że czynnikiem ją wyzwalającym jest raczej
reperfuzja, podczas gdy niedokrwienie skutkuje martwicą (nekrozą) [66,68].
Apoptoza jest ściśle kontrolowanym, ATP-zależnym procesem enzymatycznym (programowana śmierć komórki), którego końcowymi efektorami są; (i) aktywacja kaskady cysteinowych proteaz, znanych jako kaspazy, które ostatecznie degradują białka komórkowe oraz (ii)
aktywacja nukleaz (endonukleaza G oraz czynnik AIF, apoptosis-inducing factor) skutkująca
fragmentacją DNA w jądrze komórkowym (ryc. 5.13.). Do aktywacji tych enzymów i indukcji
apoptotycznej śmierci komórek mogą prowadzić trzy ścieżki sygnalizacyjne.
1. Ścieżka mitochondrialna – uruchamiana przez czynniki zwiększające przepuszczalność
błon mitochondrialnych, w tym częściowo na drodze aktywacji megakanału mitochondrialnego. Mitochondria są organellum komórkowym wyspecjalizowanym w gromadzeniu
i uwalnianiu do cytoplazmy licznych białek o działaniu apoptogennym (cytochrom c i AIF
w normalnych mitochondriach są niezbędne do prawidłowego funkcjonowania fosforylacji oksydacyjnej). Wśród nich najważniejsze to: (i) cytochrom c, białko Smac (second
mitochondria-derived activator of caspase) i proteaza HtrA2 (high temperature requirement
protein A2), które po wyjściu z mitochondriów, na drodze różnych mechanizmów, w obecności ATP, aktywują kaspazę 9 a następnie efektorową kaspazę 3 oraz (ii) endonukleaza G
i czynnik AIF, które, po przejściu do jądra komórkowego, degradują DNA w mechanizmie
niezależnym od kaspaz. Molekularny mechanizm wzrostu przepuszczalności błon mitochondrialnych i uwalniania do cytoplazmy białek pro-apoptotycznych jest słabo poznany.
104
Wiadomo, że proces ten jest ściśle regulowany, między innymi przez rodzinę cytoplazmatycznych białek Bcl-2, z których część ma działanie pro- (Bax, Bak i inne), a część antyapoptotyczne (Bcl2 i Bcl-XL). Pod wpływem czynników aktywujących apoptozę, jak ROS
czy niedokrwienie/reperfuzja, pro-apoptotyczne białka Bax i Bak tworzą por w zewnętrznej
błonie mitochondrialnej, co umożliwia uwalnianie pro-apoptotycznych białek z przestrzeni międzybłonowej. Anty-apoptotyczne białka Bcl2 i Bcl-XL inaktywują Bax i Bak, a także
zmniejszają zdolność Ca2+ do aktywacji mega-kanału mitochondrialnego i w ten sposób
zapobiegają rozszczelnianiu błon mitochondrialnych [68]. Scieżka mitochondrialna jest
prawdopodobnie szczególnie istotna w sercu ze względu na wyjątkowo dużą ilość mitochondriów w kardiomiocytach (~30% objętości komórki) oraz fakt, że mitochondria pełnią role integratora i wzmacniacza pozostałych ścieżek pro-apoptotycznych (ryc. 5.13.).
Ryc. 5.13. Uproszczony schemat procesu apoptozy w kardiomiocytach. Apoptozę mogą indukować
czynniki: (i) zewnątrzkomórkowe działające poprzez aktywację błonowego receptora śmierci; (ii)
zwiększające przepuszczalność zewnętrznej błony mitochondrialnej, co umożliwia wydostawanie się
do cytoplazmy mitochondrialnych białek pro-apoptotycznych oraz (iii) czynniki prowadzące do tzw.
stresu siateczki. Mitochondria są organellum komórkowym integrującym i wzmacniającym działanie
wszystkich ścieżek pro-apoptotycznych.
2. Ścieżka zewnątrzpochodna – uruchamiana poprzez aktywację błonowego receptora śmierci
Fas (death receptor Fas) przez białko FAS-L (Fas-ligand) obecne na zaktywowanych komórkach zapalnych; powoduje to, za pośrednictwem szeregu białek pośredniczących, aktywację kaspazy 8 a następnie efektorowej kaspazy 3, Równocześnie jednak aktywacja kaspazy 8
skutkuje zwiększonym uwalnianiem z mitochondriów cytochromu c, co skutkuje aktywacją kaspazy 9 i kaspazy 3 i wzmocnieniem sygnału zewnątrzpochodnej ścieżki apoptozy.
105
3. Ścieżka siateczki śródplazmatycznej/endoplazmatycznej; W wyniku tzw. stresu siateczki
(nagromadzenie w siateczce dużej ilości nieprawidłowo sfałdowanych białek skutkujące zaburzeniami mechanizmu gromadzenia i uwalniania Ca2+ przez siateczkę) dochodzi,
między innymi do aktywacji kaspazy 12, która następnie aktywuje efektorową kaspazę 3
i apoptozę. Wykazano, że stresowi siateczki, który, jak się okazuje, jest elementem patomechanizmu uszkodzenia niedokrwienno/reperfuzyjnego miokardium, towarzyszy także
zwiększone uwalnianie cytochromu c z mitochondriów i zwiększona aktywacja kaspazy 9.
Jest to prawdopodobnie skutkiem działania na mitochondria Ca2+ uwalnianego w nadmiarze z uszkodzonej/przepełnionej siateczki [68].
V.8.4. Mitochondrialny megakanał integratorem mechanizmów reperfuzyjnego
uszkodzenia
Pod wpływem różnych czynników, w tym zwiększonego stężeń jonów Ca2+, w wewnętrznej błonie mitochondrialnej powstaje nieselektywny kanał, przez który mogą przechodzić
do i z mitochondrium substancje o ciężarze do 1,5 kDa, w tym jony H+, Ca2+, cytochrom c
i inne. Skład białkowy tego megakanału mitochondrialnego (mPTP, mitochondrial permeability transition pore) i jego rola fizjologiczna są ciągle przedmiotem kontrowersji. Dość pewne
są natomiast następujące fakty [61,69-71]:
1. Regulatorem megakanału jest białko cyklofilina-D, związane z wewnętrzną błoną mitochondrialną. Jony Ca2+, wiążąc się z cyklofiliną-D, aktywują mega kanał a lek cyklosporyna-A, który wiąże się z cyklofiliną-D, przeciwdziała aktywacji megakanału przez Ca2+ i inne czynniki;
2. Nieodwracalna aktywacja megakanału jest końcową przyczyną reperfuzyjnego uszkodzenia miokardium. Przemawiają za tym badania eksperymentalne [69] i kliniczne [72] pokazujące, że blokowanie cyklofiliny-D (megakanału) przy pomocy cyklosporyny-A (iniekcja
cyklosporyny A tuż przed reperfuzją) redukuje wielkość martwicy zawałowej o 30–50%;
3. Z badań eksperymentalnych wynika, że do aktywacji megakanału dochodzi w pierwszych
minutach reperfuzji (izolowane serce szczura) [71] i że jest ona katastrofalna w skutkach
dla komórki gdyż skutkuje (ryc. 5.14.):
a. rozprzęganiem fosforylacji oksydacyjnej i utratą komórkowych zasobów ATP (produkcja
ATP maleje a dodatkowo mitochondria stają się konsumentem ATP powstałego w innych nieuszkodzonych mitochondriach i/lub w procesie glikolizy);
b. obrzękiem mitochondriów, pękaniem ich zewnętrznej błony i przedostawaniem się
z przestrzeni międzybłonowej do cytoplazmy cytochromu c i innych aktywatorów apoptozy (rozdz. V.8.3) oraz
c. uwalnianiem do cytoplazmy dużych ilości jonów Ca2+, co ma działanie toksyczne (rozdz.
V.7.2), między innymi skutkujące nekrozą;
4. Aktywacja mega kanału jest procesem indukowanym i wzmacnianym przez różne czynniki, którym tradycyjnie przypisywano rolę w indukowaniu reperfuzyjnego uszkodzenia
miokardium. Wśród nich najważniejsze to, związane z reperfuzją:
a. Obładowanie komórek Ca2+;
b. Zwiększona komórkowa produkcja ROS i stres oksydacyjny (tzw. reperfuzyjny paradoks
tlenowy);
c. Spadek komórkowego poziomu ATP i ADP i wysoki poziom fosforu nieorganicznego (Pi);
106
Ryc. 5.14. Centralna rola megakanału mitochondrialnego (mPTP) w mechanizmie reperfuzyjnego
uszkodzenia miokardium. Alkalizacja kardiomiocytów w reperfuzji umożliwia działanie aktywatorów
mPTP, w tym Ca2+ i ROS. Konsekwencją nieodwracalnej aktywacji mPTP jest katastrofa energetyczna
komórki a także jej apoptoza i/lub nekroza. Różne formy hartowania przeciwdziałają reperfuzyjnej aktywacji mPTP, co ma działanie kardioprotekcyjne.
d. Reperfuzyjne wypłukiwanie protonów z zakwaszonego niedokrwionego miokardium
i jego gwałtowna alkalizacja.
Każdy z wymienionych mechanizmów ma swoją indywidualną toksyczność, ale wszystkie one są także czynnikami skutkującymi aktywacją megakanału mitochondrialnego. Klasycznymi aktywatorami megakanału są czynniki wymienione w punktach a-c. Są one częściowo obecne już w czasie niedokrwienia, ale ich aktywacji megakanału zapobiega wtedy
niskie komórkowe pH. Reperfuzyjna alkalizacja miokardium usuwa ten blok a dodatkowo
reperfuzja zwiększa obecność aktywatorów megakanału;
5. Endogenne mechanizmy kardioprotekcyjne, znane jako hartowanie miokardium (rozdz.
VI.3), działają poprzez zapobieganie reperfuzyjnej aktywacji megakanału.
V.9. Śmierć komórek w niedokrwionym i reperfundowanym miokardium
V.9.1. Martwica, apoptoza, zawał – definicje
Termin „zawał mięśnia sercowego” oznacza śmierć komórek sercowych spowodowaną
niedokrwieniem, które to niedokrwienie jest wynikiem nierównowagi między przepływem
wieńcowym i zapotrzebowaniem na przepływ [73]. Śmierć kardiomiocytów towarzysząca samemu niedokrwieniu jest wynikiem głównie martwicy (necrosis). Natomiast reperfuzja prowadzi do utraty komórek częściowo w wyniku nekrozy a częściowo apoptozy.
Martwica obejmuje większe grupy komórek w danym obszarze miokardium i jest bierną
formą śmierci komórek spowodowaną niedoborem ATP, a następnie pękaniem błony komórkowej. Skutkuje to wydostawaniem się zawartości komórek do przestrzeni pozakomórkowej
i aktywacją odczynu zapalnego, co ostatecznie zapewnia uprzątanie martwych komórek przez
neutrofile i makrofagi. Białka komórkowe przedostają się częściowo do krwi krążącej, gdzie
mogą być identyfikowane jako tzw. markery białkowe zawału serca.
107
Martwica zawałowa występuje w postaci tzw. martwicy skrzepowej (coagulation necrosis),
typowej dla niereperfundowanego zawału, oraz martwicy z węzłami skurczu (contractionband necrosis) typowej dla obszarów reperfundowanych (rozdz. V.9.1). W tym ostatnim przypadku chodzi o to, że reperfuzji towarzyszy gwałtowny wzrost stężenia Ca2+ wtórny do jego
dokomórkowego napływu (rozdz. V.7) i ewentualnie uwalniania z mitochondriów (rozdz.
V.8.4.). Skutkuje to silnym i przetrwałym skurczem kardiomiocytów (bo komórkowe zasoby
ATP warunkujące rozkurcz są ograniczone), wzajemnym nadmiernym pociąganiem się sąsiadujących kardiomiocytów i wtórnym pękaniem ich błon komórkowych oraz uwalnianiem
białek komórkowych. W badaniu mikroskopowym stan ten widoczny jest jako zatarcie struktury sarkomeru i znaczne skrócenie odległości między prążkami Z kardiomiocytów.
Śmierć w wyniku apoptozy dotyczy raczej pojedynczych komórek niż ich grup, jest procesem energochłonnym, jest wynikiem zaprogramowanego „samobójstwa” komórek (rozdz.
V.8.3). Co istotne, apoptoza nie wywołuje reakcji zapalnej i nie towarzyszy jej uwalnianie białek komórkowych do krwioobiegu. Dlatego markery białkowe we krwi identyfikują ogólnie
nekrozę miokardium, niezależnie od mechanizmu w jakim powstała. Rozpoznanie apoptozy
a także rozróżnienie między martwicą skrzepową i martwicą z węzłami skurczu możliwe jest
dopiero w oparciu o różne techniki histologiczne.
Tabela 5.5. Kliniczna klasyfikacja różnych typów zawału serca
Typ 1. Spontaniczny zawał spowodowany niedokrwieniem wtórnym do takiego wydarzenia
wieńcowego jak erozja czy pęknięcie blaszki miażdżycowej. Kryterium rozpoznania:
wykazanie wzrostu a następnie spadku poziomu biomarkera powyżej średniej ± 3
odchylenia standardowe wartości w generalnej populacji (górny poziom referencyjny,
GPR) oraz obecność faktów wskazujących na obecność niedokrwienia;
Typ 2. Zawał wtórny do niedokrwienia wysiłkowego lub spoczynkowego towarzyszącego
takim stanom jak spazm naczyniowy, embolizacja tętnicy wieńcowej, anemia, arytmie,
nadciśnienie czy hipotonia;
Typ 3. Nagła śmierć sercowa, w tym zatrzymanie krążenia, często z objawami sugerującymi niedokrwienie mięśnia sercowego, ale bez weryfikacji nekrozy przy pomocy biomarkerów;
Typ 4a. Zawał serca towarzyszący pierwotnej plastyce wieńcowej (pPCI). Kryterium rozpoznania: u pacjenta z normalnym poziomem troponiny przed pPCI i (i) wzrostem biomarkera powyżej GPR – uszkodzenie około-proceduralne, (ii) wzrostem biomarkera
powyżej 3 x GPR – zawał spowodowany PCI;
Typ 4b. Zawał związany z zakrzepem w stencie udokumentowanym w arteriografii lub autopsji. Kryteria jak w Typie 4a;
Typ 5. Zawał związany z operacją pomostowania tętnicy wieńcowej (CABG). Kryterium rozpoznania: u pacjenta z normalnym poziomem troponiny przed CABG i (i) wzrostem
biomarkera powyżej GPR – uszkodzenie około-proceduralne, (ii) wzrostem biomarkera powyżej 5 x GPR – zawał zależny od CABG.
Preferowane obecnie w klinice białkowe biomarkery martwicy (nekrozy) miokardium,
cechują się prawie absolutną (cTnT, cTnI, sercowe izoformy troponiny T i troponiny I) lub
bardzo dużą (CK-MB, izoforma MB kinazy kreatynowej) specyficznością sercową i bardzo
108
wysoką czułością. Wraz z ich coraz powszechniejszym stosowaniem okazało się, że ich mniejsze lub większe uwalnianie występuje, obok „klasycznego” zawału serca, także w innych stanach, którym towarzyszy niedokrwienie miokardium, definiowane jako nierównowaga między przepływem wieńcowym i zapotrzebowaniem na przepływ (rozdz. V.2).
Wykazano ponadto, że nawet niewielkie poziomy tych biomarkerów mają niekorzystne
znaczenie rokownicze, co dowodzi ich przydatności w diagnozowaniu uszkodzenia miokardium [73,74]. Fakty te stały się powodem opracowania przez europejskie i amerykańskie towarzystwa kardiologiczne rozszerzonej definicji zawału serca (tab. 5.5.) [73].
V.9.2. Niejednorodność zaburzeń perfuzji w dorzeczu tętnicy dozawałowej
W normalnym sercu, perfuzja wieńcowa jest o ~20% większa w warstwie podwsierdziowej niż podnasierdziowej, ze względu na większe zapotrzebowanie energetyczne wsierdzia.
Wbrew potocznym opiniom, skutkiem nagłego zamknięcia tętnicy wieńcowej jest raczej
odwrócenie przezściennego profilu perfuzji w niedokrwionym obszarze, niż całkowite zatrzymanie perfuzji (ryc. 5.15.). Związane jest to z krążeniem obocznym pomiędzy tętnicami
nasierdzowymi i faktem, że przepływ pochodzący z tego krążenia jest największy w okolicy
nasierdzia i maleje w kierunku wsierdzia. Z badań eksperymentalnych (głównie na psach)
wiadomo, że stopień ograniczenia perfuzji w różnych warstwach obszaru niedokrwionego
zależy od liczby i kalibru obocznic między tętnicami epikardialnymi i że istnieje duża zmienność osobnicza pod tym względem [75]. Zmienność taka występuje także u ludzi, zwłaszcza,
że wielkość krążenia obocznego u ludzi rośnie zwykle wraz z zaawansowaniem i czasem trwania choroby wieńcowej.
Ryc. 5.15. Transmuralny profil perfuzji wieńcowej w ścianie lewej komory w normie i niedokrwieniu.
Na osi poziomej przedstawiono hipotetyczną odległość między epikardium i endokardium (grubość
ściany komory). W normie przepływ w warstwie podwsierdziowej jest większy niż w podnasierdziowej. W dorzeczu niedrożnej tętnicy wieńcowej profil perfuzji się odwraca i jest ona większa w warstwie
podnasierdziowej niż podwsierdziowej. Stopień ograniczenia przepływu jest różny w różnych sercach
w zależności od stopnia niedrożności tętnicy i wielkości krążenia obocznego.
109
Natomiast zamknięcie tętnicy nasierdziowej u zwierząt bez krążenia obocznego w krążeniu
wieńcowym (królik, szczur, niektóre gatunki świni), powoduje całkowitą utratę perfuzji we wszystkich warstwach miokardium i pełnościenny zawał po 20–30 minutach niedokrwienia [76].
Fakt, że poszczególne warstwy miokardium w dorzeczu dozawałowej tętnicy wieńcowej różnią się stopniem niedokrwienia powoduje, że ich sytuacja patofizjologiczna jest również różna.
Prace, w których oceniano wpływ przepływu pochodzącego z krążenia obocznego na losy niedokrwionego miokardium pozwoliły wyodrębnić trzy umowne stopnie niedokrwienia (ryc. 5.16.):
Ryc. 5.16. Hipotetyczny profil perfuzji wieńcowej w ścianie komory serca (ciągła linia) w dorzeczu tętnicy dozawałowej. Liniami przerywanymi zaznaczono warstwy w obrębie ściany komory, które są niedokrwione w stopniu letalnym (obszar szary, martwica w ciągu minut), krytycznym (obszar różowy,
martwica w ciągu godzin) i tolerowanym (obszar ciemnoczerwony, hibernowany). Szczegóły w tekście.
1. Niedokrwienie letalne – z rezydualną perfuzją miokardium <20%; W tych warunkach
skurcz zanika w ciągu kilkunastu sekund, a komórkowe poziomy ATP i CrP spadają do
wartości krytycznych w ciągu 20–40 minut i w tym czasie dochodzi do martwicy komórek. Obszar niedokrwiony w stopniu letalnym może uratować jedynie reperfuzja wykonana
prawie natychmiast po wystąpieniu niedokrwienia.
2. Niedokrwienie krytyczne – z 20–40% rezydualnym przepływem; W wyniku takiego niedokrwienia skurcz zanika również w ciągu sekund, natomiast dynamika wyczerpywania
się komórkowych zasobów ATP i CrP jest wolniejsza i do martwicy komórek dochodzi
w ciągu 6–12 godzin od początku niedokrwienia. Mięsień niedokrwiony krytycznie może
uratować jedynie reperfuzja. Badania eksperymentalne pokazują, że w obecności 20–30%
przepływu, martwicy może zapobiec reperfuzja następująca nie później niż w ciągu 1–2
godzin od początku niedokrwienia,. W obecności ~40% rezydualnego przepływu czas ten
wydłuża się do 6–12 godzin.
3. Niedokrwienie tolerowane – z rezydualnym przepływem >50–60%. W obecności takiego
niedokrwienia mięsień ulega hibernacji (mięsień wyłącza skurcz i w ten sposób utrzymuje
bilans energetyczny). Stan taki może utrzymywać się przez parędziesiąt godzin a następnie
110
następuje powolne umieranie komórek (rozdz. V.5.2.). Reperfuzja hibernowanego miokardium powoduje prawie natychmiastową odbudowę funkcji skurczowej mięśnia.
Niehomogenna dystrybucja perfuzji wieńcowej w dorzeczu tętnicy dozawałowej i większe zapotrzebowanie energetyczne wsierdzia sprawiają, że wsierdzie jest bardziej narażone
na uszkodzenie niż nasierdzie. W konsekwencji, w wyniku podwiązania tętnicy wieńcowej
w sercu psa, martwica zawałowa powstaje w warstwie podwsierdziowej lewej komory już po
20–40 minutach i w ciągu następnych kilku-kilkunastu godzin „wędruje” w kierunku nasierdzia (tzw. wavefront phenomenon) [77-79]. Czy i kiedy dojdzie do martwicy warstwy podwsierdziowej, jak szybka będzie wędrówka zawału w kierunku nasierdzia, oraz czy ostatecznie
powstanie zawał pełnościenny czy jedynie podwsierdziowy, jest zawsze wypadkową stopnia
niedrożności tętnicy dozawałowej i wielkości krążenia obocznego w jej dorzeczu, czyli czynników określających transmuralny profil perfuzji wieńcowej (ryc. 5.15.).
Ryc. 5.17. Zależność wielkości martwicy zawałowej od wielkości dorzecza niedrożnej tętnicy wieńcowej. Zwraca uwagę fakt, że w małych obszarach niedokrwienia może w ogóle nie dochodzić do martwicy i że im większy obszar niedokrwienia tym procentowo większa jego część umiera.
Inną ważną determinantą wielkości martwicy zawałowej jest wielkość obszaru lewej komory zaopatrywanego przez niedrożną tętnicę (ang. area at risk). Im obszar ten jest mniejszy
tym mniejsza jego frakcja ulega martwicy, a w przypadku bardzo małych obszarów (≤15%
lewej komory) do martwicy w ogóle nie dochodzi (ryc. 5.17.). Tłumaczy się to tym, że dyfuzja
zapewnia wystarczającą wymianę substancji między normalnym i niedokrwionym obszarem
miokardium, tylko w przypadku małych zawałów.
Obserwacje kliniczne dowodzą, że nasilenie powikłań, i generalnie, rokowanie u pacjentów z zawałem, zależą od wielkości martwicy zawałowej. Odkrycie, że w sercach z krążeniem
obocznym martwica zawałowa nie jest procesem jednoczasowym (jak u zwierząt bez krążenia
obocznego), ale trwającym wiele godzin [79], dało impuls do poszukiwań interwencji, które,
zastosowane na początku niedokrwienia, pomogłyby zatrzymać proces umierania komórek
i tym samym zmniejszać rozmiar martwicy. W badaniach eksperymentalnych wykazano, że
różne interwencje farmakologiczne lub metaboliczne o działaniu potencjalnie kardioprotekcyjnym jedynie zwalniają proces rozwoju martwicy zawałowej i nie wpływają na ostateczną
wielkość zawału (ryc. 5.18.). Także badania kliniczne, nie wykazały by któraś z tych interwencji działała korzystnie u pacjentów z zawałem bez reperfuzji (badania sprzed ery trombolitycznego leczenia zawału) (rozdz. VI).
111
Ryc. 5.18. Schemat pokazujący hipotetyczną dynamikę rozwoju i wielkość martwicy zawałowej (zielona
linia) i wpływ na nie interwencji kardioprotekcyjnych i reperfuzji. Lewa strona: kontrolny zawał (linia
ciągła) i kardioprotekcja (l. przerywana); Prawa strona: zawał bez reperfuzji (l. przerywana), reperfuzja
(l. czerwona) i reperfuzja z kardioprotekcją (l. brązowa). Zwraca uwagę, że reperfuzja zmniejsza wielkość zawału o ~50% i że kardioprotekcja dodana do reperfuzji zmniejsza wielkość zawału o kolejne
~50%. Martwica zawałowa ratowana przez kardioprotekcję dodaną do reperfuzji jest miarą reperfuzyjnego uszkodzenia miokardium.
Jedyną znaną interwencją ratującą niedokrwione komórki i zmniejszającą wielkość martwicy zawałowej jest repefuzja. Jednakże by była ona skuteczna, musi być wykonana na możliwie wczesnym etapie rozwoju martwicy zawałowej, kiedy żyje jeszcze możliwie dużo komórek „skazanych na śmierć zawałową” (ryc. 5.18).
V.9.3. Efekty wczesnej i późnej reperfuzji
Transmuralne zróżnicowanie niedokrwionego obszaru sprawia, że efekty reperfuzji zależą od warstwy miokardium i od długości poprzedzającego niedokrwienia. Istotę problemu
ilustruje schemat na ryc. 5.16. U hipotetycznego pacjenta rozkład perfuzji w dorzeczu dozawałowej tętnicy wieńcowej jest taki, że warstwa podwsierdziowa ściany komory jest niedokrwiona w stopniu letalnym (rezydualna perfuzja <20%), warstwa środkowa – w stopniu
krytycznym (rezydualna perfuzja 20–40%), a warstwa podnasierdziowa – w stopniu tolerowanym (rezydualna perfuzja 40–60%). Wobec tego, w godzinę od początku niedokrwienia,
warstwy podwsierdziowa, środkowa i podnasierdziowa będą, odpowiednio, w stanie martwicy, uszkodzenia (częściowo odwracalnego a częściowo nieodwracalnego) i hibernacji.
Reperfuzja wykonana w tym momencie, nie wpłynie specjalnie na warstwę podwsierdziową
i natychmiast odbuduje metabolizm i kurczliwość w warstwie podnasierdziowej. Natomiast
w warstwie środkowej komórki ulegną reperfuzyjnemu uszkodzeniu [80,81], na które składają się: szybsza śmierć nieodwracalnie uszkodzonych komórek, śmierć odwracalnie uszkodzonych komórek (komórki przeżyłyby gdyby reperfuzję przeprowadzić umiejętnie), dysfunkcja śródbłonka wieńcowego (której efektami są uszkadzający odczyn zapalny oraz no-reflow
phenomenon, ograniczający odbudowę perfuzji na poziomie mikrokrążenia, pomimo skutecznego udrożnienia tętnicy dozawałowej) oraz ogłuszenie mięśnia sercowego. W efekcie,
również w środkowej warstwie część komórek ulegnie martwicy a pozostałe, ogłuszone, będą
przez kolejne godziny, dni czy tygodnie odzyskiwały normalną kurczliwość. Im ograniczenie
112
przepływu jest większe i trwa dłużej, tym odbudowa skurczu kardiomiocytów, które przeżyły
reperfuzję jest wolniejsza. Dla przykładu, wykazano, że 5-minutowe rozdęcie balona w czasie plastyki wieńcowej powodowało 2–3 dniowy spadek funkcji skurczowej i rozkurczowej
miokardium w dorzeczu naprawianej tętnicy wieńcowej [82]. Granica między poziomem
niedokrwienia, które już prowadzi do ogłuszenia, ale jeszcze nie powoduje nieodwracalnego
uszkodzenia miokardium jest jednak bardzo nieostra.
Pozostając przy przykładzie z ryc. 5.16., jeżeli zawał tam analizowany nie będzie reperfundowany, lub reperfuzja będzie nieskuteczna, w ciągu kilku kolejnych godzin, martwica obejmie
także warstwę środkową ściany komory. Natomiast losy hibernowanej warstwy podnasierdziowej
są niepewne. Jeżeli przyjąć, że obserwacje pochodzące z modeli zwierzęcych hibernacji odnoszą
się także do patologii ludzkiej, przebieg wydarzeń może być następujący [29]. Prawdopodobnie
przez kilkanaście kolejnych godzin warstwa podnasierdziowa będzie w stanie hibernacji. Równocześnie w kolejnych paru dobach, w warstwie tej będą powstawały nowe naczynia mikrokrążenia
(angiogeneza) i naczynia krążenia obocznego (arteriogeneza) i dojdzie do normalizacji spoczynkowej perfuzji wieńcowej. Natomiast rezerwa wazodilatacyjna w tym obszarze będzie nieobecna
lub upośledzona. Wobec tego różnym sytuacjom zwiększającym obciążenie serca będą towarzyszyły powtarzające się epizody niedokrwienia i reperfuzji warstwy podnasierdziowej, prowadzące
do stanu przewlekłego ogłuszenia i postępującej degeneracji miokardium w tej warstwie (rozdz.
V.5.4.). Innymi słowy jest bardzo prawdopodobne, że u części pacjentów z zawałem podwsierdziowym i częściowo lub całkowicie niedrożną tętnicą dozawałową, warstwa podnasierdziowa, która
przeżyła zawał, będzie miała obniżoną kurczliwość, a z czasem będzie ulegać postępującej degeneracji. Uratować tę warstwę może w porę wykonane udrożnienie tętnicy dozawałowej.
Ratowanie warstwy podnasierdziowej ma prawdopodobnie ważne znaczenie praktyczne.
Bowiem odległe konsekwencje zawału, w postaci postępującej rozstrzeni, przebudowy i niewydolności lewej komory, zależą od rozległości zawału, w tym od tego czy jest on pełnościenny
czy podwsierdziowy. Nawet wąski rąbek żywego, kurczącego się mięśnia w warstwie podnasierdziowej stanowi wystarczające zabezpieczenie konstrukcji komory przed odległą szkodliwą
rozstrzenią powodowaną różnymi czynnikami mechanicznymi rozciągającymi tę konstrukcję.
W zgodzie z przedstawionym scenariuszem pozostają wyniki dużych badań klinicznych
pokazujące, że leczenie reperfuzyjne zmniejsza śmiertelność w zawale serca u ludzi [83] oraz,
że im wcześniej następuje reperfuzja tym większe jest ograniczenie rozmiarów zawału, bardziej kompletna odbudowa czynności skurczowej lewej komory serca, oraz mniejsza śmiertelność okołozawałowa [84-86].
V.9.4. Wątpliwości dotyczące dynamiki rozwoju zawału u człowieka
Warunkiem skuteczności leczenia reperfuzyjnego zawału jest znajomość dynamiki rozwoju martwicy zawałowej u ludzi. Obecna wiedza na ten temat nie pochodzi z bezpośrednich
pomiarów, ale jest „dedukowana” z badań eksperymentalnych i efektów klinicznej reperfuzji
wykonywanej w różnym czasie po zawale. Badania kliniczne wykazały, że skuteczność trombolizy w zmniejszaniu śmiertelności około zawałowej i innych powikłań zawału, jest największa
w pierwszej godzinie zawału (złota godzina) i spada do zera w ~12 godzinie (ryc. 5.19.) [87].
Utarła się wobec tego opinia, że martwica zawałowa u człowieka dokonuje się w czasie <12 godzin i standardy leczenia zawału nie rekomendują wykonywania reperfuzji po tym czasie.
113
Ryc. 5.19. Wpływ leczenia trombolitycznego zawału na śmiertelność okołozawałową. Różne wieloośrodkowe badania kliniczne (akronimy w prostokątach) dowodzą, że skuteczność trombolizy w zmniejszaniu śmiertelności jest największa w pierwszej godzinie zawału i maleje do zera między 12 i 24 godziną
od wystapienia bólu zawałowego
Następujące fakty sugerują, że zawał u człowieka rozwija się wolniej niż u psa i niż to
sugerują aktualne standardy:
1. Krążenie oboczne jest bardziej rozwinięte u człowieka niż psa, zwłaszcza u osobników
z długą historią CNS [76];
2. U pacjentów z zawałem (także STEMI) dochodzi do częstych spontanicznych rekanalizacji tętnicy dozawałowej – ~35% i ~50% przypadków, odpowiednio w czasie do 12 godzin [88] i od
12–48 godziny od początku zawału [89]. Razem wziąwszy pokazuje to, że niedokrwienie w klinice jest procesem często okresowym (vs. trwałe niedokrwienie w eksperymencie) (ryc. 5.20.);
Ryc. 5.20. Drożność tętnicy dozawałowej u 118 pacjentów z zawałem STEMI oceniana arteriograficznie
tuż przez i w 14 dni po zabiegu plastyki wieńcowej ze stentowaniem. Przed zabiegiem ~35% osób miało
normalną (TIMI-3) lub prawie normalna (TIMI-2) perfuzję wieńcową w dorzeczu tętnicy dozawałowej,
co sugeruje wczesną spontaniczną rekanalizację tej tętnicy wg [88]
3. Skuteczność reperfuzyjna trombolizy jest generalnie bardzo mała i dodatkowo dramatycznie maleje w czasie od początku zawału (2–4 godz. – 45% TIMI-3; > 6 godz. – 17% TIMI-3)
[90-92] dodatkowo częstość reokluzji po trombolizie jest większa niż po plastyce wieńcowej [90,91];
114
4. Plastyka wieńcowa góruje nad trombolizą skutecznością redukcji powikłań zawału, w tym
30-dniowej śmiertelności, niezależnie od wielkości opóźnienia terapii reperfuzyjnej w stosunku do początku zawału [85].
Razem wziąwszy sugeruje to, że dynamika rozwoju zawału u człowieka jest wolniejsza
od „dedukowanej” na podstawie klinicznych efektów trombolizy (ryc. 5.19.). Hipotezę tę
potwierdzają randomizowane badania kliniczne porównujące efekty reperfuzji przy pomocy streptokinazy i plastyki wieńcowej wykonywanych w różnym czasie od początku zawału
[92-94]. Konkluzja z tych prac jest taka, że plastyka wieńcowa, w przeciwieństwie do trombolizy, przynosi korzyści kliniczne nawet wtedy, kiedy jest stosowana po czasie dłuższym niż
12 godzin od początku zawału (ryc. 5.21.). Sugeruje to, że „okno przeżywalności” miokardium w ognisku zawałowym człowieka jest szersze niż się to obecnie sądzi (≤12 godz.), ale nie
wiadomo jak szerokie, gdyż badań klinicznych testujących przydatność kliniczną tzw. późnej
pozawałowej reperfuzji jest niewiele.
Ryc. 5.21. Wzrastająca skuteczność reperfuzji przy pomocy plastyki wieńcowej (czerwone słupki) vs.
tromboliza (niebieskie słupki) w ograniczaniu martwicy zawałowej w funkcji czasu jaki upłynął między
początkiem zawału i reperfuzją. Dane pochodzą z randomizowanego badania BRAVE II. Wielkość uratowanego obszaru przedstawiono w procentach obszaru niedokrwionego (area at risk, AaR) [89,94].
Badanie BRAVE II (ryc. 5.21.) wykazało skuteczność kliniczną plastyki wieńcowej wykonywanej między 12 i 48 godziną zawału [89,94]. Natomiast badania OAT [95] i TOSCA-2
[96] wykazały brak korzyści klinicznej reperfuzji wykonywanych między 3 i 28 dniem zawału.
Sugeruje to, że „okna przeżywalności” miokardium należy szukać gdzieś w obrębie 3 pierwszych dób zawału.
V.9.5. Pozawałowa przebudowa serca w erze leczenia reperfuzyjnego zawału
Terminem „pozawałowa przebudowa serca” (ang. remodeling) określa się zmiany strukturalne (morfologiczne) zachodzące w sercu z zawałem. Składają się to zmiany w samym ognisku zawałowym, polegające na zastępowaniu martwego miokardium tkanką łączną (wczesna
faza przebudowy). Oraz zmiany w pozostałej zdrowej części serca mające na celu dostosowanie struktury miokardium do zmienionych warunków obciążenia (późna faza przebudowy,
nieomawiana w tym opracowaniu) [97,98].
Elementami wczesnej fazy przebudowy są dwa procesy znane w literaturze angielskiej
jako infarct extension i infarct expansion.
115
Powiększanie się martwicy związane z ubytkiem żywych kardiomiocytów w strefie przejściowej między obszarem niedokrwionym i prawidłowo ukrwionym (ang. border zone) znane
jest jako infarct extension. Kardiomiocyty te umierają głównie w wyniku apoptozy. Powodem
jest nierównowaga między ich zwiększonym zapotrzebowaniem energetycznym (przejmują
obowiązki sąsiednich zmarłych kardiomiocytów) i lokalną perfuzją wieńcową. W strefie granicznej szybko rośnie gęstość nowych naczyń mikrokrążenia (angiogeneza) w wyniku zarówno „pączkowania” obecnych tam włośniczek jak i rekrutacji ze szpiku kostnego do serca komórek progenitorowych śródbłonka i tworzenia z nich nowych włośniczek. Leczenie zawału
przy pomocy tzw. komórek macierzystych, jeżeli rzeczywiście jest skuteczne, skutkuje prawdopodobnie przyspieszonym tworzeniem nowych naczyń mikrokrążenia w strefie granicznej
zawału, mniejszą apoptozą i mniejszym wymieraniem już istniejących kardiomiocytów w tej
strefie, a nie powstawaniem nowych [99].
Ryc.5.22. Bierne właściwości mechaniczne miokardium we wczesnym okresie pozawałowym. W ognisku zawału trawione są włókna mięśniowe i kolagenowe, co zmniejsza odporność tego segmentu na
rozciąganie/rozrywanie (czarna linia). Z pewnym opóźnieniem odkładane są tam włókna kolagenowe
tworzące bliznę, co odporność zwiększa (zielona linia). Istnieje wobec tego kilkudniowe okno czasowe
zmniejszonej odporności na rozciąganie (pokazane na szaro), sprzyjające powstawaniu wczesnej rozstrzeni, tętniaków i pęknięć ściany lewej komory. Okno to poszerzają (A) niesterydowe leki przeciwzapalne i glikokortykosteroidy, które opóźniają tworzenie blizny [97]. Okno to zwężają: (B) czynniki proangiogenne i komórki macierzyste przyspieszające gojenie [100] oraz (C) inhibitory metaloproteinaz
zwalniające trawienie włókien kolagenowych w martwej tkance [101].
Bierne rozciąganie przez wewnątrzkomorowe ciśnienie krwi i napięcie w ścianie komory
martwej tkanki, a następnie młodej blizny, budujących obszar zawału określane jest terminem
infarct expansion [98]. Już w kilka godzin od początku zawału, jego obszar ulega ścieńczeniu,
lewa komora się powiększa (ulega rozstrzeni), a jej kształt z eliptycznego staje się bardziej
kulisty. „Ekspansja zawału” dotyczy pierwszych dwóch tygodni zawału, gdyż później blizna
zawałowa traci podatność na rozciąganie. W ognisku zawałowym kardiomiocyty stopniowo
ulegają martwicy skrzepowej i równocześnie gromadzą się tam komórki zapalne. Naciek zapalny „wędruje” od obwodu zawału do jego centrum. Początkowo składa się głównie z granulocytów obojętnochłonnych (szczyt ich gromadzenia między 24 i 72 godziną zawału) a później
z makrofagów. Komórki te uwalniają enzymy trawiące martwe kardiomiocyty a także tkankę
116
łączną (metaloproteinazy macierzy, MMP). W efekcie, następuje uprzątanie martwej tkanki
i zastępowanie jej ziarniną, składającą się z naczyń mikrokrążenia, fibroblastów i miofibroblastów produkujących włókna kolagenowe. Gromadzenie się nowych włókien kolagenowych
następuje od 2–3 doby zawału. Wczesna blizna jest słaba i podatna na rozciąganie i dopiero
około czternastej doby osiąga sztywność podobną do obserwowanej w zdrowym miokardium.
Ostatecznie, w okresie pozawałowym istnieje kilkudniowe okno czasowe, w którym martwiczy fragment miokardium już stracił odporność na rozciąganie a młoda blizna jej jeszcze
nie nabyła (ryc. 5.22.). Jest to okres szczególnego nasilenia ekspansji zawału, co potwierdzają
obserwacje kliniczne. Dla przykładu, Weiss i wsp. [102] obserwowali 50% wzrost rozmiarów
segmentu lewej komory objętego zawałem u 30% pacjentów z pełnościennym zawałem ściany
przedniej już w ciągu pierwszego tygodnia zawału. Podobnie, powstawanie tętniaków komory i pękanie ściany lewej komory ma miejsce głównie w pierwszych dniach zawału [103].
Tabela 5.6. Cechy anatomopatologiczne zawału bez i z reperfuzją
Rozprzestrzenienie
Wygląd
Obecne komplikacje
– wstrząs kardiogenny
– „ekspansja” zawału
– tętniaki
– pęknięcia ściany
– skrzepliny przyścienne
– odczyn osierdziowy
– zawał prawej komory
Cechy histologiczne
– martwica z węzłami skurczu
– uszkodzenie małych naczyń
– krwotok śródkomórkowy
– mikrozatory w mikrokrążeniu
Zawał nie reperfundowany
transmuralne
blady, anemiczny
Zawał reperfundowany
częsty
częsta
częste
częste
częste
częsty
częsty
rzadki
rzadka
rzadkie
rzadkie
rzadkie
rzadki
rzadki
rzadka
rzadkie
rzadki
rzadkie
częsta
częste
częsty
częste
podwsierdziowe
czerwony, krwotoczny
Wraz z erą leczenia reperfuzyjnego, dramatycznie zmienił się obraz patomorfologiczny
zawału serca u człowieka (tab. 5.6.) [103]. Generalnie zawały obecnej ery są znacznie mniejsze, podwsierdziowe, „ukrwotocznione”, rzadziej powodują różnego rodzaju komplikacje
i towarzyszą im liczne zmiany naczyniowe, które z jednej strony są objawem reperfuzyjnego
uszkodzenia krążenia wieńcowego, a z drugiej prawdopodobnie stanowią substrat morfologiczny tzw. no-reflow phenomenon (rozdz. V.10).
V.10. No-reflow phenomenon
Współczesne leczenie zawału serca polega na jak najszybszym przywracaniu drożności
nasierdziowej tętnicy dozawałowej, głównie przy pomocy PCI. U części pacjentów, mimo skutecznego udrożnienia tej tętnicy, perfuzja miokardium w jej dorzeczu pozostaje upośledzona
na poziomie mikrokrążenia, co oznacza, że przynajmniej część niedokrwionych kardiomiocytów nie jest reperfundowana. Stan przywrócenia drożności dużej tętnicy i równoczesnego
braku reperfuzji mikrokrążenia określany jest terminem no-reflow phenomenon (NR).
117
Ryc. 5.23. Mechanizm no-reflow wtórny do reperfuzyjnego uszkodzenia śródbłonka wieńcowego.
Niedokrwienie i reperfuzja uszkadzają kardiomiocyty i śródbłonki. Uszkodzenie śródbłonka, poprzez
promocję adhezji i mikrozatorów we włośniczkach z udziałem leukocytów, płytek i/lub agregatów
płytkowo-leukocytarnych, uniemożliwiaja reperfuzję kardiomiocytów. Uszkodzenia kardiomiocytów
spowodowane brakiem reperfuzji oraz bezpośrednim uszkodzeniem niedokrwienno-reperfuzyjnym,
sumują się.
Wiemy obecnie, że NR (i) może dotyczyć 2/3 pacjentów z zawałem leczonych reperfuzją,
(ii) jest czynnikiem pogarszającym rokowanie oraz (iii) w części jest zjawiskiem wtórnym do
uwalniania materiału zatorowego z zatykającej skrzepliny i/lub blaszki miażdżycowej a części
do reperfuzyjnego uszkodzenia śródbłonka naczyniowego i związanego z tym odczynu zapalnego w reperfundowanym obszarze miokardium (ryc. 5.23.).
Cele leczenia zawału powinny obejmować, wobec tego, obok udrażniania tętnicy nasierdziowej, także całkowitą i trwałą reperfuzję niedokrwionych kardiomiocytów. Współczesne
terapie z sukcesem realizują cel pierwszy. Nie dysponujemy natomiast powszechnie uznanym
sposobem zapobiegania NR.
V.10.1. Aspekty kliniczne no-reflow phenomenon
Częstość występowania; Szacunki występowania NR u pacjentów z zawałem leczonych
PCI wahają się od 10% do powyżej 65% w zależności od użytej techniki pomiarowej [104].
Historycznie najstarszym, ale i najmniej czułym narzędziem jest arteriograficzna skala skuteczności reperfuzji TIMI, która, w istocie, opisuje nasierdziowy, a nie miokardialny przepływ wieńcowy (tab. 5.7.). Statystyki dotyczące wyników trombolitycznego leczenia zawału
pokazywały, że reperfuzja była w pełni skuteczna u ~53% pacjentów (TIMI 3), całkowicie
nieskuteczna (TIMI 0) u około 27% pacjentów, i że u ~20% pacjentów skutecznośc reperfuzji wynosiła 1 lub 2 w skali TIMI, co jest klasyfikowane jako NR [105-107]. W przypadku
zawałów STEMI leczonych PCI, występowanie NR mierzone skalą TIMI wynosi od 10% do
32% [104,108].
118
Tabela 5.7. Arteriograficzna skala skuteczności reperfuzji TIMI
Stopień skuteczności
„0”
„1”
„2”
„3”
Charakterystyka
Kontrast nie penetruje poza zakrzep w tętnicy dozawałowej;
Kontrast penetruje poza zakrzep ale nie wypełnia dorzecza tętnicy;
Dorzecze tętnicy wypełnia się w całości, ale wolniej niż normalnie;
Dorzecze tętnicy dozawałowej wypełnia się normalnie;
Natomiast ocena perfuzji miokardium przy pomocy arteriograficznej techniki Myocardial Blush Grade sugeruje, że występowanie NR może sięgać 50% [104,108]. Czynnościową
miarą skuteczności reperfuzji jest ocena kompletności normalizacji zmian w odcinku ST elektrokardiogramu po godzinie od PCI (ST segment resolution, STR). Ta czynnościowa technika
sugeruje, że niepełna reperfuzja jest jeszcze częstsza i występuje u 65% pacjentów z zawałem
leczonych PCI [104]. Na podobnie częste występowanie NR wskazują wstępne badania z użyciem echokardiografii kontrastowej (MCE) [109].
Odwracalność no-reflow; W kilku badaniach ocenę skuteczności reperfuzji powtarzano
kilkakrotnie w okresie po PCI. Badania są zgodne, że jedynie w około 50% przypadków NR
jest zjawiskiem trwałym. Sugeruje to, że w pozostałych 50% NR jest wynikiem zmian czynnościowych, które prawdopodobnie łatwiej poddają się leczeniu, np. przy pomocy wazodylatatorów czy leków przeciwpłytkowych [104,110].
Znaczenie rokownicze; Liczne badania pokazują, że obecność NR jest silnym czynnikiem ryzyka: (i) wczesnych powikłań zawału (arytmie, wysięk osierdziowy, tamponada serca,
wczesna niewydolność serca); (ii) niekorzystnej przebudowy lewej komory; (iii) późniejszych
wielokrotnych hospitalizacji z powodu niewydolności serca oraz (iv) zgonów. Niekorzystna
przebudowa lewej komory występuje przede wszystkim u pacjentów z utrwalonym, a nie
przemijającym NR [110].
Mechanizm; Przyczyną NR są częściowo zatory w naczyniach mikrokrążenia a częściowo obrzęk i deformacje komórek śródbłonka oraz przykurcz naczyń. Źródło materiału zatorowego nie jest do końca jasne.
W części są to prawdopodobnie mikrofragmenty skrzepliny wewnątrznaczyniowej i/lub
stenotycznej blaszki miażdżycowej uwalniane do mikrokrążenia podczas PCI. Za udziałem
tego mechanizmu przemawiają badania kliniczne REMEDIA i TAPAS pokazujące, że zabieg
PCI uzupełniony o trombektomię połączoną z aspiracją skrzepliny (zwłaszcza u osób z całkowitą okluzją tętnicy dozawałowej i dużą wewnątrznaczyniową skrzepliną) znamiennie redukuje występowanie NR [111] i skutkuje redukcją rocznej śmiertelności [112] w porównaniu
z samym PCI.
Następujące argumenty sugerują, że NR jest wtórny do niedokrwienno/reperfuzyjnego
uszkodzenia śródbłonka wieńcowego i że materiałem zatorowym, przynajmniej w części, są
komórki zapalne, płytki i/lub agregaty płytkowo-leukocytarne, a nie wyłącznie fragmenty
skrzepliny czy blaszki:
1. NR występuje w niedokrwionych/reperfundowanych narządach bez blaszek miażdżycowych i skrzeplin wewnątrznaczyniowych; Dla przykładu, pierwsza obserwacja NR pochodzi z eksperymentów na młodych, zdrowych psach bez miażdżycy, którym podwiązywano tętnicę wieńcową na 90 min [113]. NR jest częstym zjawiskiem w transplantologii.
119
Występuje w teoretycznie zdrowych narządach, które są eksplantowane (niedokrwione)
a następnie przeszczepiane (reperfuzja) (NR jest częstym powikłaniem przeszczepu nerek).
2. U zwierząt eksperymentalnych, w naczyniach mikrokrążenia miokardium w obszarze dotkniętym NR wykazano obecność granulocytów wielojądrzastych. U tych samych zwierząt,
perfuzja serca krwią bez komórek zapalnych, zapobiegała występowaniu NR.
3. Wykazano, że granulocyty mogą powodować niedrożność naczyń poprzez: (i) mechaniczne czopowanie włośniczek, których średnica jest mniejsza niż średnica granulocytów oraz
(ii) poprzez wydzielanie substancji obkurczających naczynia, jak leukotrieny i PAF.
4. U zwierząt eksperymentalnych i ludzi, w naczyniach mikrokrążenia dotkniętych NR obserwowano liczne płytki „zadherowane” do komórek śródbłonka i/lub tworzące konglomeraty
z komórkami zapalnymi [114,115]. Ciągle nie jest jednak jasne czy te zmiany płytkowe są
konsekwencją, czy przyczyną reperfuzyjnego uszkodzenia miokardium i nieskuteczności
reperfuzji. Także kliniczne próby zapobiegania płytkowo-pochodnemu uszkodzeniu miokardium u pacjentów z zawałem przyniosły niejednoznaczne wyniki [115].
5. Do NR przyczyniają się także obrzęk i skurcz samych komórek śródbłonka, powodujący
zmniejszenie światła naczyń mikrokrążenia, obserwowany podczas reperfuzji izolowanego
serca szczura (Glyn i Ward 2000).
6. Czynnikami ryzyka występowania NR są czynniki, o których wiadomo, że zwiększają
niedokrwienne (czas do reperfuzji, duży obszar niedokrwienia) i/lub reperfuzujne uszkodzenie miokardium, w tym zwiększona leukocytoza, reaktywność płytek [115] i poziomy
tromboksanu TxA2 [104] i endoteliny [116] w surowicy.
Zapobieganie i leczenie; Żadna z testowanych dotąd terapii NR nie znalazła się w standardach leczenia zawału. Najbardziej obiecujące są wspomniane powyżej wyniki badań REMEDIA i TAPAS, sugerujące skuteczność kliniczną trombektomii połączonej z aspiracją
skrzepliny. Próby farmakologicznego leczenia zakończyły się albo niepowodzeniem (adenozyna), albo, choć obiecujące, były przedmiotem jedynie małych obserwacyjnych prac wymagających dalszej weryfikacji (inhibitory receptora płytkowego IIb/IIIa, nitroprusydek sodu,
nikorandil, blokery receptora endotelinowego ET-1, antagoniści receptora tromboksanu
TxA2) [104,109].
V.10.2. Reperfuzyjna dysfunkcja śródbłonkowa
Naczynia stanowią ok. 30% tkanki sercowej i w niedokrwionym/reperfundowanym
miokardium podlegają podobnym procesom uszkadzającym jak kardiomiocyty. Szczególnie
ważne i dobrze poznane jest reperfuzyjne uszkodzenie śródbłonka wieńcowego rozwijające
się już w pierwszych paru minutach po przywróceniu przepływu. Jego manifestacjami są: (i)
dysfunkcja śródbłonkowa; (ii) destrukcja glikokaliksu komórek śródbłonka; (iii) zwiększona
przepuszczalność śródbłonka dla białek oraz (iv) zwiększona śródbłonkowa ekspresja molekuł adhezyjnych [117-121].
Dysfunkcję śródbłonkową definiuje się jako stan, w którym stwierdza się upośledzenie
czynności naczynio-rozkurczającej śródbłonka (związanej z śródbłonkową produkcją NO).
Tak stwierdzane upośledzenie produkcji NO jest markerem zaburzeń przeciwzakrzepowej,
fibrynolitycznej i przeciwzapalnej czynności śródbłonka (rozdz. IX).
120
Ryc. 5.24. Udział endoteliny (ET-1) i reaktywnych form tlenu w mechanizmie reperfuzyjnego uszkodzenia śródbłonka wieńcowego. W reperfundowanym miokardium rośnie produkcja ET-1, która aktywuje
liczne układy enzymatyczne produkujące O2–. O2– wyjątkowo aktywnie reaguje z NO, co zmniejsza biologiczną dostępność NO. Dodatkowo, toksyczny produkt reakcji, nadtlenoazotyn (ONOO–) jest czynnikiem uszkadzającym śródbłonek.
Jak pokazuje ryc. 5.24., centralną role w mechanizmie reperfuzyjnego uszkodzenia śródbłonka odgrywa reperfuzyjna nadprodukcja endoteliny i związany z tym stres oksydacyjny
[120-122]. Dodatkowo, endotelina jest substancją bardzo silnie obkurczającą naczynia krwionośne, stymuluje adhezję granulocytów, agregację płytek krwi i jest czynnikiem chemotaktycznym dla makrofagów [123], działania te mogą również przyczyniać się do reperfuzyjnego
uszkodzenia serca i/lub śródbłonka. Okazuje się, że hartowanie niedokrwieniem, zapobiega
reperfuzyjnemu uszkodzeniu śródbłonka, dlatego, że zapobiega reperfuzyjnemu wzrostowi produkcji endoteliny [122]. W badaniach klinicznych wykazano, że blokada receptorów
endotelinowych ET-1, zmniejsza występowanie NR u pacjentów z zawałem leczonych PCI
[104,109].
V.10.3. Reperfuzyjny odczyn zapalny
W okresie niedokrwienia ma miejsce bardzo powolne gromadzenie się komórek zapalnych w miokardium i proces ten dramatycznie przyspiesza w reperfuzji. Sens biologiczny tego
procesu prawdopodobnie polega na usuwaniu z ogniska zawałowego martwych komórek, co
stwarza warunki dla rozwoju procesów reparacyjnych, w tym bliznowacenia uszkodzonej
tkanki [124]. Tezę tę potwierdza wzrost częstości takich powikłań zawału, jak tętniak i pęknięcie ściany komory, u osób leczonych dużymi dawkami sterydów działającymi przeciwzapalnie. Obok korzystnych odległych efektów, naciek komórek zapalnych ma niekorzystne
działanie we wczesnym okresie reperfuzji, kiedy nasila reperfuzyjne uszkodzenie miokardium,
częściowo poprzez udział w mechanizmie NR a cześciowo poprzez cytotoksyczne działanie
121
substancji produkowanych przez aktywowane komórki zapalne. W tym kontekście wykazano, że wielkość reperfuzyjnego uszkodzenia serca koreluje z ilością granulocytów w reperfundowanym obszarze, czyli wielkością odczynu zapalnego i że interwencje zmniejszające ten
odczyn zapobiegają uszkodzeniu.
Centralną rolę w inicjacji procesu zapalnego odgrywają trzy rodzaje białek adhezyjnych
[125,126]: (i) integryny obecne częściowo na powierzchni komórek zapalnych (ale także na powierzchni płytek vide glikoproteina IIb/IIIa) a częściowo magazynowane w ziarnistościach cytoplazmatycznych; (ii) selektyny (P i E) – w normalnym śródbłonku magazynowane w ziarnistościach cytoplazmatycznych, pojawiają się na powierzchni komórek śródbłonka i płytek natychmiast po aktywacji tych komórek oraz (iii) immunoglobuliny (ICAM i VCAM), które pojawiają
się na powierzchni komórek śródbłonka ze znacznym opóźnieniem gdyż są syntetyzowane de
novo w odpowiedzi na bodziec pobudzający. Białka adhezyjne pojawiające się na powierzchni
zaktywowanych komórek śródbłonka są ligandami dla integryn i innych białek na powierzchni
komórek zapalnych i wzajemne wiązania między białkami adhezyjnymi śródbłonka i komórek
zapalnych skutkuje wychwytem tych ostatnich z krwi. Na proces ten (proces zapalny) składają się
trzy następujące po sobie zjawiska: (i) powolne toczenie się komórek zapalnych po powierzchni
śródbłonka (ang. rolling) przy udziale selektyn; (ii) adhezja czyli trwałe wiązanie komórek zapalnych ze śródbłonkiem i ich unieruchomienie przy udziale immunoglobin oraz (iii) aktywacja
komórek zapalnych, na którą składają się: zwiększona ekspresja na ich powierzchni integryn,
uwalnianie do otoczenia licznych toksycznych substancji oraz zmiany w cytoszkielecie komórek
zapalnych powodujące ich spłaszczenie, a następnie migrację (diapedezę, przy udziale immunoglobin) poprzez przestrzenie między komórkami śródbłonka do głębiej leżących tkanek.
Granulocyty naciekające mięsień sercowy a następnie aktywowane wydzielają już we
wczesnym okresie reperfuzji liczne substancje o działaniu cytotoksycznym, które zwiększają
reperfuzyjne uszkodzenie miokardium. Wśród nich najważniejsze to: (i) O2– – produkowany w dużych ilościach przez leukocytarną oksydazę NADPH i przekształcający się następnie
w H2O2; (ii) kwas podchlorawy, który jest produktem reakcji katalizowanej przez mieloperoksydazę uwalnianą z ziarnistości leukocytarnych (H2O2 + Cl– kwas podchlorawy). Kwas
podchlorawy jest silnym oksydantem, może utleniać lub chlorować różne substancje i jest
głównym czynnikiem odpowiedzialnym za cytotoksyczne działanie granulocytów zależne
od ROS; (iii) proteazy uwalniane z ziarnistości leukocytarnych, w tym: elastaza i kolagenaza
hydrolizujące białka macierzy pozakomórkowej oraz (iv) metabolity kwasu arachidonowego
(leukotrien B4,) oraz czynnik aktywujący płytki krwi (PAF). LTB4 i PAF są silnymi stymulatorami chemotaksji, adhezji i degranulacji granulocytów, co nasila ich wychwyt z krwi i zwiększa uszkodzenie tkanek spowodowane granulocytami [125].
Piśmiennictwo
1. Cohn PF, Fox KM, Daly C. Silent myocardial ischemia. Circulation 2003; 108:1263-1277.
2. Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiol Rev 2008; 88:10091086.
3. Parker JO, Chiong MA, West RO, Case RB. Sequential alterations in myocardial lactate metaboli, S-T
segments, and left ventricular function during angina induced by atrial pacing. Circulation 1969;
40:113-131.
122
4. Parker JO, West RO, Case RB, Chiong MA. Temporal relationships of myocardial lactate metabolism, left ventricular function, and S-T segment depression during angina precipitate by exercise.
Circulation 1969; 40:97-111.
5. Bin JP, Pelberg RA, Wei K, Le DE, Goodman NC, Kaul S. Dobutamine versus dipyridamole for
inducing reversible perfusion defects in chronic multivessel coronary artery stenosis. J Am Coll
Cardiol 2002; 40:167-174.
6. Selwyn AP, Forse G, Fox K, Jonathan A, Steiner R. Patterns of disturbed myocardial perfusion in
patients with coronary artery disease. Regional myocardial perfusion in angina pectoris. Circulation
1981; 64:83-90.
7. Barnes E, Hall RJ, Dutka DP, Camici PG. Absolute blood flow and oxygen consumption in stunned
myocardium in patients with coronary artery disease. J Am Coll Cardiol 2002; 39:420-427.
8. Jagathesan R, Barnes E, Rosen SD, Foale R, Camici PG. Dobutamine-induced hyperaemia inversely
correlates with coronary artery stenosis severity and highlights dissociation between myocardial
blood flow and oxygen consumption. Heart 2006; 92:1230-1237.
9. Krantz DS, Hedges SM, Gabbay FH et al. Triggers of angina and ST-segment depression in ambulatory patients with coronary artery disease: evidence for an uncoupling of angina and ischemia. Am
Heart J 1994; 128:703-712.
10. Crea F, Gaspardone A. New look to an old symptom: angina pectoris. Circulation 19971; 96:3766-3773.
11. Mega JL, Hochman JS, Scirica BM et al. Clinical features and outcomes of women with unstable
ischemic heart disease: observations from metabolic efficiency with ranolazine for less ischemia in
non-ST-elevation acute coronary syndromes-thrombolysis in myocardial infarction 36 (MERLINTIMI 36). Circulation 2010; 121:1809-1817.
12. Beltrame JF. Advances in understanding the mechanisms of angina pectoris in cardiac syndrome X.
Eur Heart J 2005; 26:946-948.
13. Lanza GA, Sestito A, Sgueglia GA et al. Effect of spinal cord stimulation on spontaneous and stressinduced angina and ‘ischemia-like’ ST-segment depression in patients with cardiac syndrome X. Eur
Heart J 2005; 26:983-989.
14. Hofkamp SE, Henrikson CA, Wegener ST. An interactive model of pain and myocardial ischemia.
Psychosom Med 2007; 69:632-639.
15. Di Monaco A, Bruno I, Calcagni ML et al. Cardiac adrenergic nerve function in patients with cardiac
syndrome X. J Cardiovasc Med (Hagerstown ) 2010; 11:151-156.
16. Gallagher KP, Osakada G, Matsuzaki M, Kemper WS, Ross J, Jr. Myocardial blood flow and function
with critical coronary stenosis in exercising dogs. Am J Physiol 1982; 243:H698-H707.
17. Fung AY, Gallagher KP, Buda AJ. The physiologic basis of dobutamine as compared with dipyridamole
stress interventions in the assessment of critical coronary stenosis. Circulation 1987; 76:943-951.
18. Calnon DA, Glover DK, Beller GA et al. Effects of dobutamine stress on myocardial blood flow,
99mTc sestamibi uptake, and systolic wall thickening in the presence of coronary artery stenoses:
implications for dobutamine stress testing. Circulation 1997; 96:2353-2360.
19. Senior R, Lahiri A. Enhanced detection of myocardial ischemia by stress dobutamine echocardiography utilizing the “biphasic” response of wall thickening during low and high dose dobutamine
infusion. J Am Coll Cardiol 1995; 26:26-32.
20. Williams RI, Payne N, Phillips T, D’hooge J, Fraser AG. Strain rate imaging after dynamic stress provides objective evidence of persistent regional myocardial dysfunction in ischaemic myocardium:
regional stunning identified? Heart 2005; 91:152-160.
21. Hauser AM, Gangadharan V, Ramos RG, Gordon S, Timmis GC. Sequence of mechanical, electrocardiographic and clinical effects of repeated coronary artery occlusion in human beings: echocardiographic observations during coronary angioplasty. J Am Coll Cardiol 1985; 5:193-197.
22. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in
health and disease. Physiol Rev 2010; 90:207-258.
23. Heusch G, Schulz R, Rahimtoola SH. Myocardial hibernation: a delicate balance. Am J Physiol 2005;
288:H984-H999.
123
24. Rahimtoola SH. The hibernating myocardium. Am Heart J 1989; 117:211-221.
25. Camici PG, Wijns W, Borgers M et al. Pathophysiological mechanisms of chronic reversible left
ventricular dysfunction due to coronary artery disease (hibernating myocardium). Circulation 1997;
96:3205-3214.
26. Depre C, Vatner SF. Mechanisms of cell survival in myocardial hibernation. Trends Cardiovasc Med
2005; 15:101-110.
27. Zhang JY, Ishibashi Y, Zhang Y et al. From AHL. Myocardial bioenergetics during acute hibernation.
Am J Physiol 1997; 42:H1452-H1463.
28. Shen YT, Kudej RK, Bishop SP, Vatner SF. Inotropic reserve and histological appearance of hibernating myocardium in conscious pigs with ameroid- induced coronary stenosis. Basic Res Cardiol 1996;
91:479-485.
29. Shen YT, Vatner SF. Mechanism of impaired myocardial function during progressive coronary
stenosis in conscious pigs - hibernation versus stunning? Circ Res 1995; 76:479-488.
30. Kudej RK, Ghaleh B, Sato N, Shen YT, Bishop SP, Vatner SF. Ineffective perfusion-contraction matching in conscious, chronically instrumented pigs with an extended period of coronary stenosis. Circ
Res 1998; 82:1199-1205.
31. Bolli R, Marban E. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev 1999;
79:609-634.
32. Camici PG, Rimoldi O. Myocardial hibernation vs repetitive stunning in patients. Cardiol Rev 1999;
7:39-43.
33. Kim SJ, Depre C, Vatner SF. Novel mechanisms mediating stunned myocardium. Heart Fail Rev
2003; 8:143-153.
34. Kloner RA, Bolli R, Marban E et al. Medical and cellular implications of stunning, hibernation, and
preconditioning: an NHLBI workshop. Circulation 1998; 97:1848-1867.
35. Heyndrickx GR, Millard RW, McRitchie RJ, Maroko PR, Vatner SF. Regional myocardial functional
and electrophysiological alterations after brief coronary artery occlusion in conscious dogs. J Clin
Invest 1975; 56:978-985.
36. Bolli R. Basic and clinical aspects of myocardial stunning. Prog Cardiovasc Dis 1998; 40:477-516.
37. Bolli R, Zughaib M, Li XY et al. Recurrent ischemia in the canine heart causes recurrent bursts of
free radical production that have a cumulative effect on contractile function - a pathophysiological
basis for chronic myocardial ‘’stunning’’. J Clin Invest 1995; 96:1066-1084.
38. Horn HR, Teichholz LE, Cohn PF, Herman MV, Gorlin R. Augmentation of left ventricular contraction pattern in coronary artery disease by an inotropic catecholamine: the epinephrine ventriculogram. Circulation 1974; 49:1063-1071.
39. Rahimtoola SH. A perspective on the three large multicenter randomized clinical trials of coronary
bypass surgery for chronic stable angina. Circulation 1985; 72 (Suppl V):V-123-V-135.
40. Ambrosio G, Betocchi S, Pace L et al. Prolonged impairment of regional contractile function after
resolution of exercise-induced angina: evidence of myocardial stunning in patients with coronary
artery disease. Circulation 1996; 94:2455-2464.
41. Gerber BL, Wijns W, Vanoverschelde JLJ et al. Myocardial perfusion and oxygen consumption in
reperfused noninfarcted dysfunctional myocardium after unstable angina - direct evidence for myocardial stunning in humans. J Am Coll Cardiol 1999; 34:1939-1946.
42. Rinaldi CA, Masani ND, Linka AZ, Hall RJ. Effect of repetitive episodes of exercise induced myocardial ischaemia on left ventricular function in patients with chronic stable angina: evidence for
cumulative stunning or ischaemic preconditioning? Heart 1999; 81:404-411.
43. Barnes E, Dutka DP, Khan M, Camici PG, Hall RJ. Effect of repeated episodes of reversible myocardial ischemia on myocardial blood flow and function in humans. Am J Physiol Heart Circ Physiol
2002; 282:H1603-H1608.
44. Vanoverschelde JL, Wijns W, Depre C et al. Mechanisms of chronic regional postischemic dysfunction in humans. New insights from the study of noninfarcted collateral-dependent myocardium.
Circulation 1993; 87:1513-1523.
124
45. Vanoverschelde JL, Wijns W, Borgers M et al. Chronic myocardial hibernation in humans. From
bedside to bench. Circulation 1997; 95:1961-1971.
46. Camici PG, Dutka DP. Repetitive stunning, hibernation, and heart failure: contribution of PET to
establishing a link. Am J Physiol 2001; 280:H929-H936.
47. Bin JP, Pelberg RA, Wei K, Coggins M, Goodman NC, Kaul S. Relation between regional function
and coronary blood flow reserve in multivessel coronary artery stenosis. Am J Physiol 2000; 279:H058-H3064.
48. Shohet RV, Garcia JA. Keeping the engine primed: HIF factors as key regulators of cardiac metabolism and angiogenesis during ischemia. J Mol Med 2007; 85:1309-1315.
49. Stanley WC, Morgan EE, Huang H et al Malonyl-CoA decarboxylase inhibition suppresses fatty acid
oxidation and reduces lactate production during demand-induced ischemia. Am J Physiol 2005;
289:H2304-H2309.
50. Chandler MP, Chavez PN, McElfresh TA, Huang H, Harmon CS, Stanley WC. Partial inhibition
of fatty acid oxidation increases regional contractile power and efficiency during demand-induced
ischemia. Cardiovasc Res 2003; 59:143-151.
51. Chandler MP, Huang H, McElfresh TA, Stanley WC. Increased nonoxidative glycolysis despite continued fatty acid uptake during demand-induced myocardial ischemia. Am J Physiol 2001; 282:
H1871-H1878.
52. Chavez PN, Stanley WC, McElfresh TA, Huang H, Sterk JP, Chandler MP. Effect of hyperglycemia
and fatty acid oxidation inhibition during aerobic conditions and demand-induced ischemia. Am J
Physiol 2003; 284:H1521-H1527.
53. Chaitman BR, Skettino SL, Parker JO et al. Anti-ischemic effects and long-term survival during ranolazine monotherapy in patients with chronic severe angina. J Am Coll Cardiol 2004; 43:1375-1382.
54. Szwed H, Sadowski Z, Elikowski W et al. Combination treatment in stable effort angiba using
trimetazidine and metoprolol. Results of a randomized, doubl blind, multicentre study (TROMPOL
II). Eur Heart J 2001; 22:2267-2274.
55. King LM, Opie LH. Glucose and glycogen utilisation in myocardial ischemia - changes in metabolism and consequences for the myocyte. Mol Cell Biochem 1998; 180:3-26.
56. Li C, Keaney JF, Jr. AMP-activated protein kinase: a stress-responsive kinase with implications for
cardiovascular disease. Curr Opin Pharmacol 2010; 10:111-115.
57. Avkiran M, Gross G, Karmazyn M, Klein H, Murphy E, Ytrehus K. Na+/H+ exchange in ischemia,
reperfusion and preconditioning. Cardiovasc Res 2001; 50:162-166.
58. Karmazyn M, Sostaric JV, Gan XT. The myocardial Na+/H+ exchanger: a potential therapeutic target
for the prevention of myocardial ischaemic and reperfusion injury and attenuation of postinfarction
heart failure. Drugs 2001; 61:375-389.
59. Karmazyn M, Moffat MP. Role of Na+/H+ exchange in cardiac physiology and pathophysiology.
Mediation of myocardial reperfusion injury by the pH paradox. Cardiovasc Res 1993; 27:915-924.
60. Hausenloy DJ, Baxter G, Bell R et al. Translating novel strategies for cardioprotection: the Hatter
Workshop Recommendations. Basic Res Cardiol 2010; 105:677-686.
61. Ovize M, Baxter GF, Di Lisa F et al. Postconditioning and protection from reperfusion injury: where
do we stand? Position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res 2010; 87:406-423.
62. Gao WD, Liu YG, Mellgren R, Marban E. Intrinsic myofilament alterations underlying the decreased
contractility of stunned myocardium - a consequence of ca2+- dependent proteolysis? Circ Res 1996;
78:455-465.
63. Gao WD, Atar D, Liu YG, Perez NG, Murphy AM, Marban E. Role of troponin I proteolysis in the
pathogenesis of stunned myocardium. Circ Res 1997; 80:393-399.
64. Hoque ANE, Haist JV, Karmazyn M. Na+-H+ exchange inhibition protects against mechanical, ultrastructural, and biochemical impairment induced by low concentrations of lysophosphatidylcholine in isolated rat hearts. Circ Res 1997; 80:95-102.
65. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med 2007; 357:1121-1135.
125
66. Wojtczak L, Zablocki K. [Mitochondria in cell life, death and disease]. Postepy Biochem 2008;
54:129-141.
67. Rouslin W, Erickson JL, Solaro RJ. Effects of oligomycin and acidosis on rates of ATP depletion in
ischemic heart muscle. Am J Physiol 1986; 250:H503-H508.
68. Lee Y, Gustafsson AB. Role of apoptosis in cardiovascular disease. Apoptosis 2009; 14:536-548.
69. Hausenloy DJ, Ong SB, Yellon DM. The mitochondrial permeability transition pore as a target for
preconditioning and postconditioning. Basic Res Cardiol 2009; 104:189-202.
70. Halestrap AP. A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem Soc Trans 2010; 38:841-860.
71. Miura T, Tanno M, Sato T. Mitochondrial kinase signalling pathways in myocardial protection from
ischaemia/reperfusion-induced necrosis. Cardiovasc Res 2010; 88:7-15.
72. Piot C, Croisille P, Staat P et al. Effect of cyclosporine on reperfusion injury in acute myocardial
infarction. N Engl J Med 2008; 359:473-481.
73. Thygesen K, Alpert JS, White HD. Universal definition of myocardial infarction. Eur Heart J 2007;
28:2525-2538.
74. Jaffe AS, Ravkilde J, Roberts R et al. It’s time for a change to a troponin standard. Circulation 2000;
102:1216-1220.
75. Reimer KA, Long JB, Murry CE, Jennings RB. Theree-dimensional distrbution of collateral blood
flow within the anatomic area at risk after circumflex coronary artery occlusin in dogs. Cardiology
1987; 82:473-485.
76. Schaper W, Gorge G, Winkler B, Schaper J. The collateral circulation of the heart. Prog Cardiovasc
Dis 1988; 31:57-77.
77. Reimer KA, Jennings RB. The “wavefront phenomenon” of myocardial ischemic cell death. II. Transmural progression of necrosis within the framework of ischemic bed size (myocardium at risk) and
collateral flow. Lab Invest 1979; 40:633-644.
78. Reimer KA, Lowe JE, Rasmussen MM, Jennings RB. The wavefront phenomenon of ischemic
cell death. 1. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation 1977;
56:786-794.
79. Jennings RB, Reimer KA. Lethal myocardial ischemic injury. Am J Pathol 1981; 102:241-255.
80. Kloner RA. Does reperfusion injury exist in humans. J Am Coll Cardiol 1993; 21:537-545.
81. Rezkalla SH, Kloner RA. No-reflow phenomenon. Circulation 2002; 105:656-662.
82. Sheiban I, Tonni S, Benussi P, Marini A, Trevi GP. Left ventricular dysfunction following transient
ischemia induced by transluminal coronary angioplasty. Beneficial effects of calcium antagonists
against post-ischemic myocardial stunning. Eur Heart J 1993; 14 (Suppl):14-21.
83. Yusuf S, Sleight P, Held P, McMahon S. Routine medical menagement of acute myocardial infarction: lessons from overviews of recent randomized controlled trials. Circulation 1990; 82 (Suppl.
II):II-117-II-134.
84. Gibson CM. Time is myocardium and time is outcomes. Circulation 2001; 104:2632-2634.
85. Boersma E. Does time matter? A pooled analysis of randomized clinical trials comparing primary
percutaneous coronary intervention and in-hospital fibrinolysis in acute myocardial infarction patients. Eur Heart J 2006; 27:779-788.
86. Brodie BR, Stone GW, Cox DA et al. Impact of treatment delays on outcomes of primary percutaneous coronary intervention for acute myocardial infarction: analysis from the CADILLAC trial. Am
Heart J 2006; 151:1231-1238.
87. Newby K. Clinical outcomes according to time to treatment. Clin Cardiol 1997; 20:III11-III15.
88. Ndrepepa G, Kastrati A, Schwaiger M et al. Relationship between residual blood flow in the infarctrelated artery and scintigraphic infarct size, myocardial salvage, and functional recovery in patients
with acute myocardial infarction. J Nucl Med 2005; 46:1782-1788.
89. Schomig A, Mehilli J, Antoniucci D et al. Mechanical reperfusion in patients with acute myocardial infarction presenting more than 12 hours from symptom onset: a randomized controlled trial.
JAMA 2005; 293:2865-2872.
126
90. Becker RC. Reocclusion following successful thrombolysis - emerging concepts. Cardiology 1993;
82:265-273.
91. Verheugt FWA, Meijer A, Lagrand WK, Vaneenige MJ. Reocclusion: the flip side of coronary
thrombolysis. J Am Coll Cardiol 1996; 27:766-773.
92. Schomig A, Ndrepepa G, Kastrati A. Late myocardial salvage: time to recognize its reality in the
reperfusion therapy of acute myocardial infarction. Eur Heart J 2006; 27:1900-1907.
93. Zijlstra F, Patel A, Jones M et al. Clinical characteristics and outcome of patients with early (<2 h),
intermediate (2-4 h) and late (>4 h) presentation treated by primary coronary angioplasty or thrombolytic therapy for acute myocardial infarction. Eur Heart J 2002; 23:550-557.
94. Schomig A, Ndrepepa G, Mehilli J et al. Therapy-dependent influence of time-to-treatment interval
on myocardial salvage in patients with acute myocardial infarction treated with coronary artery
stenting or thrombolysis. Circulation 2003; 108:1084-1088.
95. Hochman JS, Lamas GA, Buller CE et al. Coronary intervention for persistent occlusion after myocardial infarction. N Engl J Med 2006; 335:1-13.
96. Dzavik V, Buller CE, Lamas GA et al. Randomized trial of percutaneous coronary intervention for subacute infarct-related coronary artery occlusion to achieve long-term patency and improve ventricular
function: the Total Occlusion Study of Canada (TOSCA)-2 trial. Circulation 2006; 114:2449-2457.
97. Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 1990; 81:1161-1172.
98. Mitchell GF, Lamas GA, Vaughan DE, Pfeffer MA. Left ventricular remodeling in the year after first
anterior myocardial infarction: A quantitative analysis of contractile segment lengths and ventricular shape. J Am Coll Cardiol 1992; 19:1136-1144.
99. Kocher AA, Schuster MD, Szabolcs MJ et al. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and
improves cardiac function. Nat Med 2001; 7:430-436.
100. Minatoguchi S, Takemura G, Chen XH et al. Acceleration of the healing process and myocardial regeneration may be important as a mechanism of improvement of cardiac function and remodeling by
postinfarction granulocyte colony-stimulating factor treatment. Circulation 2004; 109:2572-2580.
101. Jugdutt BI. Ventricular remodeling after infarction and the extracellular collagen matrix: when is
enough enough? Circulation 2003; 108:1395-1403.
102. Weiss JL, Marino PN, Shapiro EP. Myocardial infarct expansion: recognition, significance and pathology. Am J Cardiol 1991; 68:35D-40D.
103. Basso C, Rizzo S, Thiene G. The metamorphosis of myocardial infarction following coronary recanalization. Cardiovasc Pathol 2010; 19:22-28.
104. Niccoli G, Burzotta F, Galiuto L, Crea F. Myocardial no-reflow in humans. J Am Coll Cardiol 2009;
54:281-292.
105. Ito H, Maruyama A, Iwakura K et al. Clinical implications of the “no refow” phenomenon. A predictor of complications and left ventricular remodeling in reperfused anterior wall myocardial
infarction. Circulation 1996; 93:223-228.
106. Kaul S, Ito H. Microvasculature in acute myocardial ischemia: part II: evolving concepts in
pathophysiology, diagnosis, and treatment. Circulation 2004; 109:310-315.
107. Kaul S, Ito H. Microvasculature in acute myocardial ischemia: part I: evolving concepts in
pathophysiology, diagnosis, and treatment. Circulation 2004; 109:146-149.
108. Rezkalla SH, Dharmashankar KC, Abdalrahman IB, Kloner RA. No-reflow phenomenon following
percutaneous coronary intervention for acute myocardial infarction: incidence, outcome, and effect of pharmacologic therapy. J Interv Cardiol 2010; 23:429-436.
109. Ito H. No-reflow phenomenon in patients with acute myocardial infarction: its pathophysiology
and clinical implications. Acta Med Okayama 2009; 63:161-168.
110. Galiuto L, Lombardo A, Maseri A et al. Temporal evolution and functional outcome of no reflow: sustained and spontaneously reversible patterns following successful coronary recanalisation.
Heart 2003; 89:731-737.
127
111. Burzotta F, Trani C, Romagnoli E et al. Manual thrombus-aspiration improves myocardial reperfusion: the randomized evaluation of the effect of mechanical reduction of distal embolization by
thrombus-aspiration in primary and rescue angioplasty (REMEDIA) trial. J Am Coll Cardiol 2005;
46:371-376.
112. Svilaas T, Vlaar PJ, van dH, I, Diercks GF et al. Thrombus aspiration during primary percutaneous
coronary intervention. N Engl J Med 2008; 358:557-567.
113. Kloner RA, Ganote CE, Jennings RB. The “no-reflow” phenomenon after temporary coronary occlusion in the dog. J Clin Invest 1974; 54:1496-1508.
114. Michaels AD, Gibson CM, Barron HV. Microvascular dysfunction in acute myocardial infarction:
focus on the roles of platelet and inflammatory mediators in the no-reflow phenomenon. Am J
Cardiol 2000; 85:50B-60B.
115. Barrabes JA, Inserte J, Agullo L, Alonso A, Mirabet M, Garcia-Dorado D. Microvascular thrombosis: An exciting but elusive therapeutic target in reperfused acute myocardial infarction. Cardiovasc
Hematol Disord Drug Targets 2010; 10:273-283.
116. Niccoli G, Lanza GA, Shaw S et al. Endothelin-1 and acute myocardial infarction: a no-reflow mediator after successful percutaneous myocardial revascularization. Eur Heart J 2006; 27:1793-1798.
117. Deedwania PC. Endothelium: a new target for cardiovascular therapeutics. J Am Coll Cardiol 2000;
35:67-70.
118. Pernow J, Gonon AT, Gourine A. The role of the endothelium for reperfusion injury. Eur Heart J
Supplements 2004; 3 (Supplement C):C22-C27.
119. Beresewicz A, Czarnowska E, Maczewski M. Ischemic preconditioning and superoxide dismutase
protect against endothelial dysfunction and endothelium glycocalyx disruption in the postischemic
guinea-pig hearts. Mol Cell Biochem 1998; 186:87-92.
120. Maczewski M, Beresewicz A. The role of endothelin, protein kinase C, and free radicals in the
mechanism of the post-ischemic endothelial dysfunction in guinea-pig hearts. J Mol Cell Cardiol
2000; 32:297-310.
121. Kurzelewski M, Czarnowska E, Beresewicz A. Endothelin in the mechanism of endothelial injury
and neutrophil adhesion in the post-ischemic guinea pig heart. Eur J Pharmacol 2002; 434:95-107.
122. Duda M, Konior A, Klemenska E, Beresewicz A. Preconditioning protects endothelium by preventing ET-1-induced activation of NADPH oxidase and xanthine oxidase in post-ischemic heart. J Mol
Cell Cardiol 2007; 42:400-410.
123. Kirkby NS, Hadoke PW, Bagnall AJ, Webb DJ. The endothelin system as a therapeutic target in cardiovascular disease: great expectations or bleak house? Br J Pharmacol 2008; 153:1105-1119.
124. Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction.
Cardiovasc Res 2002; 53:31-47.
125. Jordan JE, Zhao ZQ, Vinten-Johansen J. The role of neutrophils in myocardial ischemia-reperfusion
injury. Cardiovasc Res 1999; 43:860-878.
126. Molteni R, Fabbri M, Bender JR, Pardi R. Pathophysiology of leukocyte-tissue interactions. Curr
Opin Cell Biol 2006; 18:491-498.
128
VI. Kardioprotekcja
Andrzej BERĘSEWICZ
Termin „kardioprotekcja” jest współcześnie definiowany głównie jako ograniczanie niedokrwienno/reperfuzyjnego uszkodzenia miokardium (V.1). Wyróżnia się (ryc. 6.1.):
Ryc. 6.1. Schemat dwóch typów kardioprotekcji (szczegóły w tekście).
1. kardioprotekcję bezpośrednią, w której substancje o spodziewanym działaniu protekcyjnym podaje się przed, w trakcie, lub po niedokrwieniu, z założeniem, że ich korzystne
działanie dotyczy jedynie okresu, w którym mają one kontakt z miokardium.
2. kardioprotekcję w mechanizmie „hartowania” miokardium – wykorzystywany jest tu endogenny mechanizm kardioprotekcyjny polegający na tym, że pod wpływem pewnych
bodźców (np. krótkotrwałe nieuszkadzające niedokrwienie) w miokardium pozostaje „pamięć” w postaci przedłużającego się w czasie okresu zwiększonej tolerancji miokardium na
niedokrwienie/reperfuzję. Jeżeli kolejny incydent niedokrwienia/reperfuzji trafi na okres
zwiększonej tolerancji, spowodowane nim uszkodzenie będzie mniejsze. Pierwsze próby
zastosowania różnych form hartowania w klinice są dość obiecujące (rozdz. VI.3).
129
Era intensywnych poszukiwań sposobów ograniczania konsekwencji niedokrwienia/reperfuzji zaczęła się w 1974 roku, kiedy to Braunwald i Maroko [1], w oparciu o obserwacje,
że rokowanie pacjentów z zawałem pogarsza się wraz z ilością utraconego miokardium, zaproponowali hipotezę, że sposobem na poprawę rokowania mogłoby być leczenie nastawione
na ograniczanie rozmiaru martwicy zawałowej. Mniej więcej w tym samym czasie wykazano,
że rozwój martwicy zawałowej jest procesem rozciągniętym w czasie (zaczyna się w warstwie
podwsierdziowej i powoli wędruje w kierunku nasierdzia) [2,3] (ryc. 5.18.), co pokazało, że
istnieje dość długi przedział czasowy, w którym proponowane przez Braunwalda i Maroko
leczenie kardioprotekcyjne mogłoby być skuteczne. W kolejnych latach przebadano tysiące
interwencji o potencjalnym działaniu kardioprotekcyjnym, głównie w różnych zwierzęcych
modelach zawału. W 1980 roku ukazała się publikacja DeWood’a i wsp. [4] dowodząca, że
przyczyną większości zawałów jest niedrożność tętnicy zawałowej spowodowana skrzepliną.
W efekcie nastąpiła era trombolitycznego leczenia zawału i fascynacji leczeniem reperfuzyjnym i jego konsekwencjami.
Dotychczasowe badania na temat kardioprotekcji można podsumować w następujących
punktach:
1. W modelach eksperymentalnych, interwencje farmakologiczne lub metaboliczne o działaniu kardioprotekcyjnym jedynie zwalniają proces rozwoju martwicy zawałowej i nie wpływają na ostateczną wielkość zawału (ryc. 5.18.). Podobnie, brak dowodów klinicznych by
któraś z tych interwencji działała korzystnie u pacjentów z zawałem bez reperfuzji (badania
sprzed ery trombolitycznego leczenia zawału). Teoretyzując, leczenie opóźniające proces
umierania mogłoby znaleźć zastosowanie kliniczne także w dobie reperfuzyjnego leczenia
zawału. Opóźnianie rozwoju martwicy powinno, bowiem skutkować wydłużeniem czasu
po zawale serca, w którym reperfuzja jest jeszcze skuteczna. Hipoteza ta wymaga jednak
weryfikacji klinicznej.
2. Reperfuzja, przeprowadzona odpowiednio wcześnie, jest jedyną znaną interwencją ratującą niedokrwione komórki przed śmiercią, więc zgodnie z definicją, jest interwencją o działaniu kardioprotekcyjnym. Zgodnie z hipotezą Braunwalda i Maroko, reperfuzja poprawia
rokowanie pacjentów z zawałem, jeżeli tylko skutkuje redukcją wielkości zawału.
3. Ostateczny wynik reperfuzji jest za każdym razem sumą jej korzystnych i niekorzystnych
efektów. Innymi słowy, spodziewany korzystny efekt reperfuzji jest zwykle pomniejszany
o skutki reperfuzyjnego uszkodzenia miokardium (ryc. 5.18.).
4. W modelach zwierzęcych wykazano, że różne interwencje kardioprotekcyjne nacelowane na
„walkę” tylko z reperfuzyjnym uszkodzeniem (interwencje przeciw-zapalne, przeciw-wolnorodnikowych, zapobiegające komórkowej akumulacji Ca2+, hartowanie i inne), zmniejszają
martwicę w niedokrwionym/reperfundowanym sercu nawet o 30–50% (ryc. 5.18.).
5. Badania kliniczne testujące możliwości farmakologicznego ograniczania reperfuzyjnego
uszkodzenia serca w kontekście OZW zakończyły się spektakularnym niepowodzeniem.
Powody tego mogą być liczne, ale generalnie nie są zrozumiałe [5-7]. W efekcie w USA
i Europie nie zarejestrowano dotąd żadnego leku przeznaczonego do ograniczania wielkości zawału u pacjentów z OZW.
6. Jedynymi pozytywnym przykładami skutecznej kardioprotekcji w klinice są kardioplegia
w kardiochirurgii (rozdz. VI.1.) i ewentualnie skuteczność leków „wieńcowych” w lecze130
niu stabilnej choroby wieńcowej (rozdz. VI.2). Jedna z interpretacji tego faktu jest taka,
że kardioprotekcja farmakologiczna może być skuteczna u ludzi jedynie w przypadkach
łagodnego i/lub krótkotrwałego niedokrwienia.
7. Ograniczanie reperfuzyjnego uszkodzenia serca pozostaje ciągle ważnym aczkolwiek niezrealizowanym celem terapeutycznym i z pewnością będzie przedmiotem dalszych intensywnych badań. W tej sytuacji ostrożne nadzieje budzą wstępne doniesienia o możliwości
praktycznego wykorzystania w klinice endogennych mechanizmów kardioprotekcyjnych,
w tym hartowania (rozdz. VI.3).
VI.1. Krążenie pozaustrojowe i transplantacja serca
Elementem obu procedur jest czasowe zatrzymanie krążenia wieńcowego, co jest równoznaczne z wprowadzeniem serca w stan niedokrwienia. Kilkugodzinną skuteczną ochronę
serca przed uszkodzeniem niedokrwienno/reperfuzyjnym uzyskuje się w tych stanach poprzez drastyczne ograniczenie zapotrzebowania energetycznego miokardium. Służą do tego
dwa postępowania:
1. Kardioplegia (paraliż serca), która polega na całkowitym wyłączeniu czynności skurczowej
miokardium, o której wiadomo, że zużywa ok. 80% całkowitej energii produkowanej przez
serce. W tym celu łożysko krążenia wieńcowego jest wypełniane jednym ze znanych płynów
kardioplegicznym (np. wg receptury Bretschneidera lub wg St. Thomas Hospital), których
najważniejszym składnikiem jest chlorek potasu w stężeniu ~20 mEq/l. W obecności tak
zwiększonego stężenia K+, potencjał spoczynkowy kardiomiocytów rośnie od normalnej
wartości około – 85 mV do potencjału powyżej – 60 mV, przy którym kardiomiocyty stają
się niepobudliwe elektrycznie i wobec tego się nie kurczą.
2. Hipotermia w postaci tzw. zimnej kardioplegii i/lub oziębiania całego ciała pacjenta – drastycznie ogranicza potrzeby energetyczne związane z podstawowymi procesami życiowymi
miokardium. Obowiązuje tutaj zasada, że redukcja temperatury miokardium o 10oC powoduje redukcje aktualnej MVO2 o 50%. Połączenie kardioplegii z hipotermią jest w stanie
zredukować MVO2 nawet o 97%.
Kardioplegia, sama lub z hipotermią, jest przykładem postępowania kardioprotekcyjnego
o wyjątkowej skuteczności. Jednakże nawet radykalne ograniczenie wydatku energetycznego,
uzyskiwane w wyniku kardioplegii, nie zapewnia trwałego zabezpiecza miokardium przed
uszkadzającymi konsekwencjami przedłużającego się niedokrwienia. Jeżeli serce ma podjąć
normalną funkcję, prędzej czy później musi być poddane reperfuzji. W istocie, kardioplegia opóźnia jedynie, a nie przeciwdziała, rozwojowi uszkodzenia w niedokrwionym miokardium.
VI.2. Stabilna choroba wieńcowa
Niedokrwienie w przebiegu stabilnej choroby wieńcowej występuje wtedy, kiedy przepływ wieńcowy (zaopatrzenie miokardium w O2) nie nadąża za aktualnymi potrzebami, co
skutkuje utratą równowagi energetycznej komórek sercowych (rozkład ATP jest większy niż
jego produkcja) (rozdz. V.2.2). W konsekwencji incydenty niedokrwienia w stabilnej chorobie
131
wieńcowej są na ogół łagodne i krótkotrwałe i skutkują głównie dolegliwościami bólowymi
ograniczającymi zdolność wykonywania wysiłku (angina), lokalnymi zaburzeniami kurczliwości miokardium, ewentualnie zaburzeniami rytmu i jedynie rzadko – śmiercią komórek.
Główne cele leczenia stabilnej choroby wieńcowej to: (i) prewencja OZW oraz (ii) poprawa
jakości życia pacjentów poprzez zapobieganie i/lub łagodzenie incydentów niedokrwienia
wysiłkowego i ich konsekwencji, takich jak ogłuszenie. Istnieją dobrze udokumentowane wieloośrodkowe badania wykazujące, że ten drugi cel można osiągnąć przy pomocy interwencji
farmakologicznych poprawiających równowagę energetyczną miokardium poprzez:
1. poprawę prefuzji wieńcowej (β-blokery, iwabradyna, azotany, blokery kanału wapniowego);
2. zmniejszanie obciążenia energetycznego serca (β-blokery, iwabradyna, azotany, blokery
kanału wapniowego);
3. poprawę efektywności mechanicznej mięśnia sercowego poprzez częściowe hamowanie komórkowego utleniania NEFA i „przekierowywanie” przy pomocy tzw. modulatorów metabolizmu przemian energetycznego serca ze spalania NEFA na spalanie glukozy. W efekcie mniej ATP jest trwonione na różne Ca2+–zależne reakcje niezwiązane bezpośrednio ze
skurczem;
4. kombinację powyższych trzech postępowań.
VI.2.1. β-blokery i iwabradyna
Badania epidemiologiczne dowodzą, że osoby z szybką czynnością serca żyją krócej [8].
Z drugiej strony, dobrze udokumentowany jest fakt, że β-blokery: (i) poprawiają tolerancję wysiłku u pacjentów ze stabilną chorobą wieńcową, zwłaszcza tych ze stenozą wieńcową klasy II
(zwężenie ograniczające średnice tętnicy nasierdziowej o 50–80%); (ii) zmniejszają śmiertelność osób z chorobą wieńcową po zawale mięśnia sercowego lub ze skurczową niewydolnością
serca oraz (iii) że nasilenie tych korzystnych efektów jest proporcjonalne do stopnia zwolnienia
czynnności serca [9]. Wobec mnogości różnych sercowych efektów β-blokady, mechanizm tych
działań jest niepewny. Najczęściej dyskutowane są następujące mechanizmy:
1. Zapobieganie ostrym zespołom wieńcowym. Najczęstszą przyczyną OZW jest nagłe przerwanie ciągłości śródbłonka i/lub pokrywy łącznotkankowej pokrywających blaszkę miażdżycową, co prowadzi do powstawania zatykającej tętnicę skrzepliny w miejscu uszkodzenia blaszki [10]. Ważną przyczyną uszkodzenia blaszki jest prawdopodobnie fala tętna
rytmicznie rozciągająca ścianę tętnic i ewentualnie powodująca mechaniczne rozerwanie
tkanki pokrywającej blaszkę. Przyjmuje się, że β-blokery zmniejszają występowanie OZW
gdyż, redukując czynność serca i/lub ciśnienie tętnicze, zmniejszają prawdopodobieństwo
mechanicznego uszkodzenia blaszek miażdżycowych. Można wobec tego oczekiwać, że
każda interwencja zwalniająca czynność serca powinna zmniejszać występowanie OZW
w chorobie wieńcowej.
2. Zapobieganie incydentom niedokrwienia wysiłkowego na drodze przynajmniej trzech mechanizmów. β-blokada:
a. poprzez zwolnienie czynności serca, działanie inotropowo ujemne oraz ewentualnie obniżenie ciśnienia tętniczego, zmniejsza zapotrzebowanie tlenowe serca, i w ten sposób
poprawia równowagę energetyczną miokardium; Wiadomo, że 50% przyspieszenie czynności serca powoduje około 50% wzrost konsumpcji tlenu przez serce;
132
b. zmniejsza zapotrzebowanie tlenowe nasierdzia w obszarze za stenozą oraz w zdrowych
obszarach komory i w ten sposób zmniejsza podkradanie krwi z warstwy podwsierdziowej do podnasierdziowej w obszarze za stenozą;
c. wraz ze zwolnieniem czynności serca, rośnie długość fazy rozkurczu i efekt ten jest szczególnie duży przy rytmach bliskim fizjologicznym [11] (ryc. 3.4.) co skutkuje spadkiem
oporu kompresyjnego w obszarze za stenozą i jego lepszą perfuzją. W kontekście działania przeciw-niedokrwiennego wykazano u psów ze stenozą wieńcową, że β-blokada
(atenolol) rzeczywiście zapobiega, związanej z wysiłkiem, niekorzystnej redystrybucji
przepływu wieńcowego z wsierdzia do nasierdzi i towarzyszącej utracie kurczliwości
miokardium w obszarze za stenozą [12] i że te korzystne efekty atenololu nie występują
u zwierząt, u których utrzymywano stałą częstość pracy serca poprzez stymulację przedsionków [13]. Razem wziąwszy, badania te sugerują, że β-blokada zapobiega wysiłkowemu niedokrwieniu mięśnia sercowego głównie poprzez zwalnianie czynności serca.
Tabela 6.1. Niepożądane efekty leków wieńcowych
Antagoniści β-receptorów
• Upośledzenie kurczliwości serca
• Zaburzenia przewodnictwa A-V
• Skurcz tętnic nasierdziowych w sąsiedztwie
ekscentrycznej blaszki miażdżycowej i/lub
spazm naczyniowy
• Skurcz oskrzeli
• Chromanie u osób z miażdżycą zarostową
tętnic obwodowych
• Nadwrażliwość na β-stymulację po
odstawieniu β-blokera
• Hiperlipidemia
• Cukrzyca
• Depresja
• Zaburzenia potencji u mężczyzn
Antagoniści kanału wapniowego
o Upośledzenie kurczliwości serca
o Zaburzenia przewodnictwa A-V
o Zaparcia
o Obrzęki obwodowe
o Bóle głowy
o Zaburzenia widzenia
o Rozwój tolerancji na leki blokujące kanał
wapniowy
W zgodzie z tą hipotezą, iwabradyna, która wybiórczo zwalnia czynność serca i nie ma
innych efektów hemodynamicznych, ma działanie przeciw-niedokrwienne analogiczne do
β-blokady [14,15]. Iwabradyna nie ma natomiast działań niekorzystnych typowych dla tradycyjnych „leków wieńcowych” (tab. 6.1.). Szczególnie istotny jest tu prawdopodobnie fakt,
że w wyniku β-blokady może dochodzić do prowokowanego wysiłkiem skurczu lub spazmu
tętnic wieńcowych w okolicach ekscentrycznych blaszek miażdżycowych (co ogranicza antyniedokrwienny efekt β-blokady) a także do chromania u osób z miażdżycą zarostową tętnic
obwodowych, których to niekorzystnych efektów nie ma iwabradyna. Chodzi o to, że wysiłkowi
fizycznemu towarzyszy proporcjonalny wzrost aktywności współczulnego układu nerwowego.
Mediator tego układu, noradrenalina, działa na tętnice poprzez receptory α zlokalizowane na
komórkach mięśni gładkich budujących ścianę tętnic oraz receptory β zlokalizowane na komórkach śródbłonka naczyń. Aktywacja receptorów α powoduje skurcz tętnic, a receptorów
β – ich rozkurcz. W normalnych tętnicach efekty te znoszą się nawzajem i aktywacja współczulna ma niewielki wpływ na regulację oporu i przepływu wieńcowego (i ewentualnie przepływu
133
w kończynach dolnych). Sytuacja zmienia się, kiedy czynność śródbłonka jest upośledzona (jak
w przypadku miażdżycy tętnic wieńcowych czy miażdżycy zarostowej) lub, kiedy receptory β na
śródbłonku są zablokowane. Wtedy wysiłkowi fizycznemu (aktywacji współczulnej) może towarzyszyć paradoksalny przykurcz tętnic nasierdziowych (i/lub obwodowych). W przeciwieństwie
do β-blokady, iwabradyna nie ogranicza wysiłkowego wzrostu przepływu wieńcowego.
Jeżeli chodzi o farmakologiczny mechanizm działania iwabradyny [16,17,18]. Fizjologicznym rozrusznikiem serca są komórki węzła zatokowego. Za ich automatyzm odpowiedzialne
są cztery prądy jonowe, w tym prąd rozrusznikowy IF (płynący przez kanał F), którego zahamowanie skutkuje zwolnieniem rytmu zatokowego. O iwabradynie wiadomo, że [15,18]:
1. Jest selektywnym i specyficznym (blokuje IF w stosunkowo niskich stężeniach 0,1–100 μM)
antagonistą IF i w tym mechanizmie zwalnia rytm zatokowy serca;
2. Jej siła blokowania IF rośnie wraz z częstością pracy serca;
3. Nawet, jeżeli całkowicie zahamuje IF, zwalnia czynność serca jedynie o ok. 15 uderzeń/min,
zarówno w spoczynku jak i w czasie wysiłku [14];
4. Nie wpływa na przewodzenie w węźle A-V ani na inne parametry elektrofizjologiczne komórek sercowych;
5. Nie wpływa na kurczliwość mięśnia sercowego, poza wpływem, jaki wynika ze zwolnienia
częstości akcji serca;
6. Ostatecznie, na poziomie narządowym, jej działanie polega na wybiórczym (selektywnym)
zwalnianiu czynności serca, bez wpływu na inne aspekty pracy serca.
VI.2.2. Blokery kanału wapniowego
Leki te są antagonistami kanałów wapniowych typu L, które w układzie krążenia występują w:
1. kardiomiocytach – ich zablokowanie zmniejsza dokomórkowy napływ Ca2+ w czasie potencjału czynnościowego, co skutkuje zmniejszeniem siły skurczu mięśnia sercowego;
2. węźle zatokowym, gdzie biorą udział w spoczynkowej depolaryzacji – ich blokowanie zwalnia depolaryzację i częstotliwość rytmu serca;
3. węźle przedsionkowo-komorowym – ich blokowanie zwalnia lub blokuje przewodnictwo A-V;
4. komórkach mięśni gładkich naczyń wieńcowych – ich blokowanie zmniejsza dokomórkowy napływ Ca2+, co skutkuje rozkurczem naczyń. W sercu ten ostatni efekt jest szczególnie
widoczny w dużych tętnicach nasierdziowych.
Blokery kanału wapniowego pochodne dihydropirydyny (np. amlodypina) działają głównie na komórki mięśni gładkich naczyń powodując rozkurcz tętnic wieńcowych i obwodowych. Natomiast niedihydropirydynowe blokery kanału wapniowego (np. werapamil, diltiazem) działają na wszystkie wymienione typy komórek, więc dodatkowo zmniejszają częstość
akcji serca, szybkość przewodzenia w węźle A-V oraz kurczliwość mięśnia sercowego.
Na przeciwdławicowe działanie blokerów kanału wapniowego składają się prawdopodobnie następujące mechanizmy:
1. Rozkurcz/zapobieganie przykurczowi (w tym spazmowi) dużych tętnic wieńcowych: mechanizm szczególnie istotny w przypadku dławicy Prinzmetala, zaburzeń mikrokrążenia
i dolegliwości dławicowych pochodzących od ekscentrycznych blaszek miażdżycowych;
2. Zmniejszanie zapotrzebowania tlenowego mięśnia sercowego (tylko niedihydropirydynowe) poprzez zmniejszenie częstotliwości rytmu serca i obniżenie kurczliwości mięśnia ser134
cowego. Ten mechanizm bardzo przypomina działanie β-blokerów i rozumowanie przedstawione w rozdziale VI.2.1 ma i tutaj zastosowanie.
VI.2.3. Nitrogliceryna i inne organiczne azotany
Głównym reprezentantem organicznych azotanów jest nitrogliceryna. Inne leki z tej grupy to dwuazotan izosorbidu, monoazotan izosorbidu oraz czteroazotan pentaerytritolu. Azotany są powszechnie używane w leczeniu stabilnej i niestabilnej choroby wieńcowej, ostrego
zawału, przewlekłej niewydolności serca, obrzęku płuc i nadciśnienia tętniczego. Korzystne anty-niedokrwienne działanie azotanów związane jest z faktem, że leki te równocześnie
zmniejszają zapotrzebowanie miokardium na O2 oraz poprawiają perfuzję miokardium, co
ostatecznie skutkuje poprawą równowagi energetycznej miokardium i mniejszym niedokrwieniem [19,20].
Następujące efekty decydują o tym, że azotany redukują zapotrzebowania mięśnia sercowego na O2:
1. Małe stężenia azotanów rozkurczają w sposób preferencyjny duże żyły i tętnice. W konsekwencji krew gromadzi się w krążeniu trzewnym i maleje powrót żylny krwi do płuc
i serca. Maleją: ciśnienie napełniania komór, naprężenie w ich ścianach i ogólnie obciążenie
wstępne serca. Rzut minutowy zdrowego serca w obecności azotanów może maleć. Natomiast rzut skurczowy i minutowy pacjentów z upośledzoną funkcją lewej komory mogą się
lekko zwiększać;
2. W obecności większych dawek leków i/lub ich wyższych stężeń we krwi następuje także
rozkurcz tętniczek. Skutkuje to zmniejszeniem oporu naczyniowego i obciążenia następczego lewej komory;
3. Wraz z redukcją obciążenia wstępnego i ewentualnie następczego maleje zapotrzebowanie
miokardium na O2, co skutkuje poprawą bilansu energetycznego miokardium i w efekcie
ogranicza lub likwiduje niedokrwienie.
Efekty decydujące o tym, że azotany poprawiają perfuzję wieńcową to:
1. Redystrybucja perfuzji wieńcowej z warstwy podnasierdziowej do podwsierdziowej miokardium; angina wysiłkowa polega na tym, że w obszarze zaopatrywanym przez zwężoną
tętnicę wieńcową, wraz z obciążeniem serca, perfuzja miokardium w warstwie podnasierdziowej rośnie kosztem redukcji perfuzji w warstwie podwsierdziowej (efekt podkradania), co skutkuje niedokrwieniem głównie warstwy podwsierdziowej (ryc. 3.1.). Azotany
zmniejszają lub zapobiegają tej niekorzystnej redystrybucji przepływu wieńcowego;
2. Zapobieganie lub łagodzenie lokalnego skurczu tętnicy wieńcowej, spontanicznego lub sprowokowanego wysiłkiem fizycznym, w sąsiedztwie ekscentrycznej blaszki miażdżycowej;
3. Rozkurcz naczyń krążenia obocznego;
4. Działanie antyagregacyjne na płytki krwi.
Azotany są pro-lekami, które w wyniku enzymatycznej bioaktywacji w ścianie naczyniowej (w komórkach mięśni gładkich i śródbłonka), uwalniają substancję czynną jaką jest
tlenek azotu (NO) i ewentualnie S-nitrosothiol. Powstały w ten sposób NO, ze względu na
jego bardzo krótki okres półtrwania, działa lokalnie, głównie w miejscu powstawania. NO
pochodzący z azotanów działa w podobny sposób i uzupełnia działanie NO produkowanego
przez śródbłonek. Działanie azotanów jest jednak niezależne od śródbłonka.
135
NO pochodzący z biotransformacji azotanów i śródbłonkowy działają poprzez aktywację
rozpuszczalnej cyklazy guanylowej, zwiększoną produkcję cGMP i ostatecznie aktywację kinazy białkowej G i rozkurcz mięśni gładkich (ryc. 6.2., prawa strona).
Ryc. 6.2. Efekty biologiczne związane z biotransformacją nitrogliceryny (GTN). Prawa strona: Mitochondrialna dehydrogenaza alkoholowa (mtALDH) uwalnia z GTN tlenek azotu (NO), który aktywuje
kolejno: rozpuszczalną cyklazę guanylową (sGC), produkcję cGMP i kinazę białkową G (PKG), co skutkuje rozkurczem naczyń. W ten sam sposób działa NO produkowany przez śródbłonkową syntazę NO
(eNOS) pod wpływem shear stress i agonistów. Lewa strona: Równocześnie, GTN stymuluję produkcję
O2– przez mitochondrialny łańcuch oddechowy. To skutkuje oksydacją i inaktywacją mtALDH oraz
aktywacją naczyniowej oksydazy NADPH i niekorzystnymi efektami wywołanymi przez O2– produkowany przez ten enzym.
Za bioaktywację nitrogliceryny i pentaeritritolu (ale chyba nie pozostałych organicznych
azotanów) do NO, odpowiedzialna jest mitochondrialna izoforma dehydrogenazy alkoholowej (mtALDH). Równocześnie, nitrogliceryna działając na mitochondrialny łańcuch oddechowy, zwiększa mitochondrialną produkcję O2–, który z kolei aktywuje naczyniową oksydazę
NADPH (ryc. 6.2., lewa strona) [20-23]. O2– i naczyniowy stres oksydacyjny związany z aktywacją oksydazy NADPH mają szereg niekorzystnych efektów towarzyszących przewlekłemu
podawaniu azotanów. Najważniejsze to: (i) inaktywacja NO pochodzenia śródbłonkowego,
a także inaktywacja śródbłonkowej syntazy NO, skutkujące dysfunkcją śródbłonkową; (ii)
zmniejszona dostępność i/lub produkcja NO w centralnym układzie nerwowym skutkująca
przesunięciem równowagi współczulno/przywspółczulnej na korzyść aktywacji współczulnej
136
(NO produkowany w mózgu hamuje ośrodki współczulne w rdzeniu przedłużonym) oraz
(iii) inaktywacja mtALDH, skutkująca upośledzeniem biotransformacji nitrogliceryny co razem z inaktywacją NO jest przyczyną tzw. tolerancji na azotany.
mtALDH jest głównym enzymem odpowiedzialnym za usuwanie aldehydu octowego
(przekształca aldehyd w kwas octowy), głównego toksycznego produktu utleniania alkoholu. Około 40% populacji wschodniej Azji jest nosicielem mutacji genu mtALDH skutkującej zmniejszoną aktywnością enzymu, znacznie obniżoną tolerancją alkoholu oraz obniżoną
efektywnością działania nitrogliceryny [24,25]. Z drugiej strony, równoczesne przyjmowanie
organicznych azotanów i konsumpcja alkoholu skutkują objawami analogicznymi do występujących pod wpływem alkoholu u alkoholików leczonych disulfiranem (anticol, esperal),
który jest inhibitorem mtALDH. Dodatkowo w pracy na zdrowych ochotnikach wykazano,
że disulfiran wywołuje dysfunkcję śródbłonkową i tolerancję na nitroglicerynę [26]. Razem
wziąwszy badania te pokazują, że mtALDH jest enzymem odpowiedzialnym za biotransformacje organicznych azotanów także u ludzi.
Sumując, azotyny stosowane doraźnie mają liczne doraźne korzystne efekty anty-niedokrwienne. Stosowane przewlekle rozwijają działania, które są potencjalnie niekorzystne (naczyniowy stres oksydacyjny, dysfunkcja śródbłonkowa, zwiększona aktywność współczulna).
Jednakże, znaczenie prognostyczne przewlekłego podawania azotynów w stabilnej choroby wieńcowej nie było nigdy badane w sposób systematyczny. Interesujące, że nie udało się
wykazać by podawanie azotanów we wczesnym okresie zawału miało jakikolwiek wpływ na
przeżywalność pacjentów [19].
VI.2.4. Modulatory metabolizmu
W przeciwieństwie do „klasycznych leków wieńcowych”, modulatory nie upośledzają skurczu mięśnia sercowego ani hemodynamiki serca. Stąd oczekiwanie, że będą cennym
uzupełnieniem standardowego leczenia stabilnej choroby wieńcowej i potencjalnie innych
postaci CNS. Eksperymenty zwierzęce wykazały, że substancje te zwiększają efektywność mechaniczną i tempo reperfuzyjnej odbudowy czynności skurczowej niedokrwionego mięśnia
sercowego, nawet wtedy, kiedy są podawane tuż przed lub tuż po rozpoczęciu reperfuzji, co
oznacza, że punktem uchwytu ich działania jest reperfuzyjny mechanizm ogłuszenia [27,28].
Wśród potencjalnie korzystnych efektów hamowania utleniania NEFA, najważniejsze jest
prawdopodobnie odhamowywanie dehydrogenazy pirogronianowej (PDH) (tab. 6.2.). Dostępne są trzy strategie terapeutycznego hamowania utleniania NEFA:
1. Zapobieganie wzrostowi i/lub obniżanie poziomu NEFA we krwi, co utrudnia ich dokomórkowy transport i dalsze spalanie w kardiomiocytach skutkujące hamowaniem PDH przez
produkty β-oksydacji (rozdz. IV.6); Poziomy noradrenaliny we krwi rosną w ciągu kilku
minut niedokrwienia (angina, OZW, krążenie pozaustrojowe) i pozostają podwyższone
nawet przez kolejnych kilka godzin, zależnie od ciężkości niedokrwienia. Katecholaminy,
poprzez aktywację β-receptorów i hormonozależnej lipazy (HLP) w tkance tłuszczowej,
zwiększają uwalnianie NEFA do krwi i równocześnie zmniejszają trzustkowe uwalnianie
insuliny, co dodatkowo aktywuje HLP (rozdz. IV.6). W efekcie ekspozycja kardiomiocytów
na NEFA w niedokrwieniu/reperfuzji rośnie, co może skutkować zwiększonym hamowaniem PDH, kwasicą komórkową i toksyczną akumulacją Ca2+ (rozdz. V.7). Korzystne efekty
137
β-blokerów w różnych postaciach CNS mają prawdopodobnie związek, między innymi,
z faktem, że w wyniku ich działania maleje poziom NEFA we krwi i metabolizm serca
przestawia się częściowo na zwiększone spalanie glukozy [27]. Kwas nikotynowy skutecznie obniża poziom NEFA we krwi, ale jest źle tolerowany. Mieszanina glukoza + insulina
+ potas (GIK), poprzez hamowanie HLP (rozdz. V.6), zmniejsza poziom NEFA we krwi
i w modelach eksperymentalnych zmniejsza niedokrwienno/reperfuzyjne uszkodzenie serca. U ludzi, u których stosowano GIK jako dodatek do kardioplegii, pooperacyjny powrót
funkcji skurczowej był szybszy i bardziej kompletny a częstość migotania przedsionków
mniejsza [29], jednakże wyniki te wymagają potwierdzenia w badaniu wieloośrodkowym.
Natomiast GIK zastosowany jako uzupełnienie leczenia reperfuzyjnego zawału u ludzi (infuzja tuż przed lub dopiero w reperfuzji) nie miał korzystnego działania [30-32].
Tabela 6.2. Potencjalne mechanizmy kardioprotekcyjnego działania interwencji hamujących
spalanie NEFA
• Koszt tlenowy produkcji ATP zmniejszony o 12%
• Mniejsze rozprzeganie fosforylacji oksydacyjnej
• Odhamowanie dehydrogenazy pirogronianowej (PDH)
o mniejsza kwasica komórkowa
o mniejsza komórkowa akumulacja Ca2+
o mniejsze trwonienie ATP
o zwiększona efektywność mechaniczna miokardium
• Inny nieznany mechanizm?
2. Hamowanie transportu NEFA do mitochondriów poprzez hamowanie CPT-I, enzymu biorącego udział w transporcie NEFA z cytoplazmy do macierzy mitochondrialnej (V.4). Badane są trzy inhibitory CPT-I: etomoxir, oxfenicina i perhexelina. Substancje te zwiększają
utlenianie glukozy i działają anty-niedokrwiennie w modelach eksperymentalnych. Wstępne wyniki badań klinicznych z etomoxirem i oxfenicyną są zachęcające, ale substancje te
mają wiele efektów niepożądanych. Perheksylina – lek „wieńcowy” z lat 70. i 80. ubiegłego
wieku, przestał być używany ze względu na działania uboczne. Obecnie zainteresowanie
lekiem wraca gdyż okazało się, że jest skutecznym inhibitorem CPT-I, i że w małych nietoksycznych dawkach wciąż ma działanie anty-anginalne [27,28].
3. Hamowanie spalania NEFA poprzez hamowanie β-oksydacji NEFA. W Europie, jako lek anty-anginalny w stabilnej chorobie wieńcowej zarejestrowany jest bloker β-oksydacji trimetazydyna, a w USA podobna strukturalnie substancja – ranolazyna (rejestracja w 2006 r.).
W modelach eksperymentalnych oba leki przestawiają metabolizm serca na spalanie glukozy
i działają kardioprotekcyjnie w niedokrwionym/reperfundowanym miokardium. Randomizowane badania kliniczne wykazały skuteczne działanie anty-anginalne trimetazydyny
(TACT, TRIMPOL II) i ranolazyny (MARISA, CARISA ERICA, MERLIN-TIMI36) zarówno
w monoterapii jak i w kombinacji z klasycznymi lekami wieńcowymi [27,28,33-35]. Ranolazyna ma także działanie antyarytmiczne, co ma prawdopodobnie związek z blokowaniem
przez nią tzw. późnego kanału sodowego i związaną z tym mniejszą komórkową akumulacją
Ca2+. Kanał ten, nieobecny w zdrowych komórkach sercowych, jest elementem przebudowy
elektrycznej miokardium w niewydolności serca i przewlekłym migotaniu przedsionków.
138
VI.3. Hartowanie serca
VI.3.1. Definicje
Hartowanie jest formą kardioprotekcji z wykorzystaniem endogennych mechanizmów
kardioprotekcyjnych (ryc. 6.1.).
W 1986 roku Murry i wsp. [36] wykonali słynny eksperyment na uśpionych psach, w którym w grupie kontrolnej podwiązywali zwierzętom tętnicę wieńcową na 40 min a w grupie
badanej 40 minutowe niedokrwienie poprzedzali czterema 5 minutowymi epizodami niedokrwienia rozdzielonymi 5 min okresami reperfuzji (ryc. 6.3.A i B). Wykazali, że te krótkie
poprzedzające niedokrwienia/reperfuzje skutkują drastyczną redukcją martwicy zawałowej.
W kontroli i grupie badanej zawał stanowił, bowiem odpowiednio 30% i 7% obszaru niedokrwionego, mimo że niedokrwienie w grupie badanej trwało w sumie 50% dłużej. Autorzy nazwali to zjawisko ischemic preconditioning (ang. conditioning oznacza indukowanie
odruchu warunkowego). W polskiej literaturze funkcjonuje termin hartowanie (serca) niedokrwieniem (HN).
Ryc. 6.3. Schemat klasycznego protokołu hartowania: (B), niedokrwieniem (ischemic preconditioning)
oraz (C) reperfuzją (ischemic postconditioning). Szczegóły w tekście.
W roku 2003, w eksperymencie na psach, Zhao i wsp. [37] wykazali, że podobne działanie ograniczające wielkość zawału jak HN ma także hartowanie reperfuzją (HR, ischemic
postconditioning), tj. zabieg, w którym miokardium poddawane jest działaniu krótkich incydentów niedokrwienia i reperfuzji (w tym wypadku 3 niedokrwienia a 30 sek w odstępach 30
sekundowych) na początku reperfuzji, a nie przed niedokrwieniem (ryc. 6.3.C).
Wykazano ponadto, że krótkie hartujące zamknięcie tętnicy pozasercowej (ramiennej,
udowej etc.) skutkuje zwiększoną odpornością miokardium na uszkodzenie niedokrwienno/
reperfuzyjne. Zjawisko to znane jest jako hartowanie na odległość (remote preconditioning,
preconditioning at a distance).
W różnych modelach eksperymentalnych wykazano, że trening fizyczny ma efekty sercowe
analogiczne do HN (exercise preconditioning, hartowanie wysiłkiem fizycznym) [38].
W przypadku HN i HR wiadomo, że zabiegi te redukują martwicę w niedokrwionym/
reperfundowanym miokardium poprzez zapobieganie reperfuzyjnej aktywacji megakanału
mitochondrialnego i następnie reperfuzyjnej komponencie uszkodzenia (ryc. 5.14.) [39].
139
Najlepiej scharakteryzowane zostało HN, o którym wiemy, że [40-42]:
1. jest endogennym i do tego niezwykle silnym mechanizmem cytoprotekcyjnym;
2. występuje u wszystkich badanych dotąd ssaków, w tym i u człowieka;
3. dotyczy, obok miokardium, także innych narządów (mózg, nerka, żołądek, oko, wątroba);
4. obok 4 epizodów niedokrwienia/reperfuzji (vide klasyczny eksperyment Murry i wsp.),
wywołują je także inne bodźce wytrącające miokardium (lub inne narządy) z równowagi
energetycznej, w tym:
• nawet pojedynczy epizod niedokrwienia trwający 1,5–2,5 minuty;
• 15 min częściowa okluzja tętnicy wieńcowej;
• krótkie okresy szybkiej stymulacji serca nie wywołującej niedokrwienia;
• krótkie okresy niedotlenienia;
• krótkie epizody przeciążenia objętościowego komory;
• 15 min pobyt w środowisku o temperaturze 42oC (heat shock);
5. redukcja martwicy zawałowej nie jest jedynym korzystnym efektem HN. Inne to [41]:
• redukcja wielkości martwicy zawałowej;
• wolniejszy spadek poziomu ATP i fosfokreatyny w niedokrwionym miokardium;
• mniej zaburzeń rytmu w czasie niedokrwienia i reperfuzji;
• szybsza i bardziej kompletna odbudowa skurczu mięśnia w czasie reperfuzji;
• redukcja apoptozy w niedokrwionym i reperfundowanym miokardium;
• mniejsze reperfuzyjne uszkodzenie śródbłonka naczyń wieńcowych;
• mniejsza ekspresja cząstek adhezyjnych na śródbłonku i mniejsza adhezja granulocytów
w niedokrwionym i reperfundowanym miokardium;
• mniejszy zakrzep w naczyniach wieńcowych zależny od płytek krwi.
6. kardioprotekcja związana z HN ma przebieg dwufazowy (ryc. 6.4.).
Ryc. 6.4. Dwie fazy zwiększonej odporności miokardium na uszkadzające działanie niedokrwienia i reperfuzji spowodowane hartowaniem. Faza wczesna rozpoczyna się prawie natychmiast po bodźcu hartującym,
trwa 1–2 godziny i jest spowodowana aktywacją tzw. kinaz białkowych wczesnej protekcji. Faza późna rozpoczyna się 24 godz. po bodźcu, trwa 3 doby i jest spowodowana aktywacją „genów późnej protekcji”.
140
VI.3.2. Molekularny mechanizm hartowania
Od dawna podejmowane są próby zastąpienia inwazyjnej i dość skomplikowanej technologicznie procedury HN jakąś formą hartowania farmakologicznego (zastąpienie hartowania
lekiem). Skuteczność tych usiłowań jest jednak ograniczona brakiem powszechnie akceptowanej hipotezy na temat molekularnego mechanizmu HN. Trudność wynika z faktu, że
w mechanizmie HN zaangażowanych jest kilka równoległych ścieżek sygnalizacyjnych i gatunki różnią się stopniem udziału poszczególnych ścieżek w mechanizmie kardioprotekcji.
Szczupłość informacji dotyczących serca ludzkiego sprawia, trudno dzisiaj spekulować, które
zwierzę jest najbliższe człowiekowi, jeżeli chodzi o mechanizm HN.
Zgodnie z aktualną hipotezą, na komórkowy mechanizmu HN składają się następujące
elementy [43-47] (ryc. 6.5.):
Ryc. 6.5. Uproszczony schemat ścieżek sygnalizacyjnych odpowiedzialnych za wczesną i późną fazę
hartowania niedokrwieniem. Szczegóły w tekście.
1. W wyniku hartującego niedokrwienia z miokardium uwalniają się substancje czynne wyzwalające proces hartowania, w tym adenozyna (działająca poprzez receptory A1 i A3),
opioidy (via receptor δ1), bradykinina (via receptory B2) oraz noradrenalina (głównie
w sercu szczura, via receptory α1);
2. Substancje te, działając na odpowiednie receptory błonowe, inicjują wewnątrzkomórkowe
ścieżki sygnalizacyjne prowadzące do aktywacji kanału potasowego zlokalizowanego w wewnętrznej błonie mitochondrialnej regulowanego poziomem ATP (mitochondrialny kanał
potasowy ATP-zależny, mitoK-ATP), która to aktywacja skutkuje niewielką mitochondrialną produkcją O2– i innych ROS. Substancjami aktywującymi mitoK-ATP i naśladującymi
działanie HN są diazoksyd i pinacidyl. Glibenklamid i inne pochodne sulfonylo-mocznika
blokują mitoK-ATP i efekty HN;
3. Mitochondrialne ROS aktywują ścieżki sygnalizacyjne, których efektorami są:
a. Kaskada wewnątrzmitochondrialnych kinaz efektorowych (kinazy wczesnej protekcji,
kinazy RISK, Reperfusion Injury Salvage Kinases). Aktywacja RISK skutkuje „uszczelnieniem” megakanału mitochondrialnego w reperfuzji (kinazy zapobiegają reperfuzyjnej
aktywacji megakanału), co stanowi ważny argument na rzecz hipotezy o centralnej roli
megakanału w mechanizmie reperfuzyjnego uszkodzenia miokardium. Ważnymi elemen141
tami RIASK są: kinaza Akt, kinaza Erk ½, kinaza 3β syntazy glikogenu (GSK-3β) oraz
enzym glikolityczny heksokinaza (HK). Końcowymi efektorami kaskady są najpewniej
HK i GSK-3β i ten ostatni enzym prawdopodobnie fosforyzuje jakiś niezidentyfikowany
białkowy komponent megakanału, skutkujący jego zmniejszoną wrażliwością na czynniki
aktywujące. Aktywacja RISK może się okazać atrakcyjnym celem działania interwencji
kardioprotekcyjnych. Wskazuje na to fakt, że do aktywacji RISK dochodzi (przynajmniej
w modelach eksperymentalnych) w wyniku działania takich interwencji kardioprotekcyjnych jak hartowanie niedokrwieniem, hartowanie reperfuzją oraz podawanie razem z reperfuzją insuliny, erytropoetyny, peptydów natriuretycznych czy statyn [44].
b. Geny licznych białek, w tym indukowalnej syntazy tlenku azotu (iNOS) i wtórnie do
tego – mitochondrialnej izoformy dyzmutazy ponadtlenkowej (MnSOD). Zwiększona
ekspresja tych genów skutkuje zwiększoną ochroną anty-rodnikową kardiomiocytów, co
jest prawdopodobnie ważnym mechanizmem późnej fazy kardioprotekcji [48].
VI.3.3. Hartowanie w sercu człowieka
Na obecność mechanizmu hartowania w sercu człowieka wskazują: (i) badania eksperymentalne na fragmentach miokardium pobranych śródoperacyjnie lub z serc eksplantowanych; (ii) obserwacje sugerujące, że bóle wieńcowe, które sygnalizują fakt niedokrwienia miokardium, paradoksalnie zwiększają tolerancję serca na kolejne dłuższe niedokrwienia oraz
(iii) nowe obserwacyjne prace kliniczne sugerujące skuteczność hartowanie niedokrwieniem,
hartowania reperfuzją i hartowania na odległość w ograniczaniu tzw. okołoproceduralnego
uszkodzenia miokardium oraz martwicy zawałowej.
VI.3.3.1. Warm up phenomenon
U osób z chorobą wieńcową nasilenie anginy wysiłkowej, związanej z codzienną aktywnością, maleje w miarę upływu dnia i powtarzania wysiłków. Zjawisko to znane jest w literaturze
anglosaskiej jako warm up phenomenon (zjawisko „rozgrzewki”) lub walk-through angina.
VI.3.3.2. Dwie następujące po sobie próby wysiłkowe
W bardziej zobiektywizowanej postaci hartowanie można zademonstrować porównując
wyniki dwóch kolejnych prób wysiłkowych u osób z chorobą wieńcową. Tolerancja drugiego
testu jest z reguły lepsza (mniejsze zmiany ST, dłuższy czas do wystąpienia zmian ST i bólu
etc.) zarówno wtedy, kiedy drugi test następuje 30 minut [49-51], jak i 24–72 godziny po
pierwszym [52]. Prawdopodobna interpretacja tych obserwacji jest taka, że niedokrwienie towarzyszące pierwszemu testowi wysiłkowemu miało działanie hartujące z klasyczną wczesną
(obecną po 30 min) i późną fazą kardioprotekcji (obecną po 24–72 godzinach).
VI.3.3.3. Angina przed zawałem
Pacjenci z bólami wieńcowymi w okresie poprzedzającym zawał serca, w porównaniu
z osobami bez poprzedzającej anginy, lepiej tolerują zawał. Dla przykładu, w badaniu TIMI 4,
występowanie zgonów szpitalnych, wstrząsu i niewydolności serca było mniejsze u pacjentów
z anginą przed zawałem. Również wielkość martwicy zawałowej, mierzona wielkością uwalniania CPK, była mniejsza u pacjentów z anginą przed zawałem. Obie grupy nie różniły się ilością
142
i rodzajem przyjmowanych leków i miały podobnie rozwinięte krążenie oboczne. Średni czas
między wystąpieniem bólu zawałowego i wdrożeniem leczenia trombolitycznego był istotnie
dłuższy u pacjentów z anginą przed zawałem, co powinno działać na niekorzyść tej grupy pacjentów, a mimo to jej stan kliniczny był lepszy [53]. Zgodnie z hipotezą, że bóle wieńcowe
mają działanie hartujące, badanie TIMI 9 wykazało, że pacjenci, którzy mieli bóle wieńcowe
w okresie 24 godzin przed zawałem, mieli w okresie 30 dni po zawale mniej takich wydarzeń
sercowych jak śmierć sercowa, powtórny zawał, niewydolność serca i wstrząs niż grupa pacjentów z anginą we wcześniejszym okresie przed zawałem, lub w ogóle bez anginy [54].
VI.3.3.4. Hartowanie u pacjentów z zawałem
Hartowanie niedokrwieniem (HN) jest mało przydatne w leczeniu pacjentów z klasycznym zawałem gdyż wymaga zastosowania bodźca hartującego w okresie poprzedzającym
niedokrwienie (ryc. 6.3.B). Idea wykorzystania mechanizmu hartowania w leczeniu zawału
odżyła z chwilą odkrycia, że hartowanie reperfuzją (HR) (ryc. 6.3.C) jest równie skuteczne jak
HN i że molekularny mechanizm obu tych hartowań jest identyczny.
Ukazało się dotąd kilka prac obserwacyjnych (tab. 6.3.) obejmujących pacjentów z zawałem STEMI, u których, w trakcie zabiegu plastyki wieńcowej ze stentowaniem, wywoływano 3
lub 4 krótkotrwałe (30–60 sek) zamknięcia tętnicy dozawałowej, naśladujące klasyczny zabieg
HR (ryc. 6.3.C). Wszystkie te prace donoszą o korzystnym efekcie HR w postaci zmniejszenia
wielkości martwicy zawałowej (mierzonego uwalnianiem enzymów wskaźnikowych), apoptozy i/lub poprawy frakcji wyrzucania w obserwacji odległej [55].
Tabela 6.3. Badania kliniczne testujące efekt hartowania reperfuzją (HR) u pacjentów z zawałem STEMI poddanych zabiegowi plastyki wieńcowej ze stentowaniem
Czas do PCI (godz.) Protokół
Efekt
K/HR
HR
Staat, 2005 [56]
5,5/5,3
4 x 60 s
CK-MB (72 h)
Ma, 2006 [57]
<12
3 x 30 s
CK-MB (72 h)
qWMSI (8 tyg.)
Darling, 2007 [58] 59/56
55/62
<12
4 rozpręż. max CK
Thibault, 2008 [59] 21/17
56/56
4,9/4,7
4 x 60 s
qperfuzja SPECT
qLVEF po 12 m.
Zhao, 2009 [60]
26/24/25 63/58/59 4,4/4,8/5,8
3 x 60 s
TnT, Apoptoza
3 x 30 s
qLVEF
Autor
N
K/HR
16/14
47/47
Wiek
K/HR
56/58
64/64
Alternatywną strategię testowali niedawno Piot i wsp. [61]. Autorzy ci, zamiast indukować
mechanizm kardioprotekcji poprzez hartowanie, zbadali efekt farmakologicznego zablokowania reperfuzyjnej aktywacji megakanału, czyli procesu, o którym wiadomo, że jest blokowany
przez hartowanie. Użyli w tym celu cyklosporyny-A, która obok działania immunosupresyjnego, blokuje cyklofilinę-D – białko biorące udział w aktywacji megakanału mitochondrialnego (V.8.4). Do badania włączono pacjentów z zawałem STEMI z potwierdzoną niedrożnością tętnicy dozawałowej (TIMI 0). Kontrolni pacjenci byli poddani tylko plastyce wieńcowej
ze stentowaniem a grupa badana tuż przez zabiegiem otrzymywała dodatkowo jednorazową
iniekcję dożylną cyklosporyny A (2,5 mg/kg). Okazało się, że cyklosporyna-A zmniejszyła
wielkość martwicy zawałowej mierzonej wielkością uwalniania CK-MB o ~40%.
143
Konkludując, obserwacyjne badania kliniczne pokazują, że zarówno HR jak i farmakologiczny „mimetyk” hartowania skutkują podobną redukcją reperfuzyjnego uszkodzenia miokardium u pacjentów z zawałem STEMI. Kliniczna przydatność tych obiecujących obserwacji
wymaga jednak weryfikacji w dużych wieloośrodkowych badaniach.
VI.3.3.5. Hartowanie w okołoproceduralnym uszkodzeniu miokardium
Planowe plastyki wieńcowe oraz operacje kardiochirurgiczne z udziałem krążenia pozaustrojowego są procedurami, którym towarzyszy planowane, okresowe niedokrwienie mięśnia sercowego i związane z tym uszkodzenie miokardium. Objawia się ono, między innymi,
obecnością we krwi biomarkerów martwicy miokardium (CK-MB, Troponiny) i jest określane terminem „około-proceduralne uszkodzenie miokardium” (periprocedural myocardial
injury) [62]. Dla przykładu, u około 30% pacjentów z planowaną plastyką wieńcową stwierdza się podwyższone poziomy biomarkerów, a u 15% z nich wzrosty te są tak duże, że upoważniają do rozpoznania okołoproceduralnego zawału serca (vide Typ 4a i Typ 5 zawału,
Tab. 5.5.). Mechanizm takiego uszkodzenia towarzyszącego plastyce wieńcowej wyjaśnia
ryc. 6.6. Znaczenie okołoproceduralnych uszkodzeń podkreśla fakt, że proporcjonalnie do wielkości uwalniania markerów martwicy miokardium pogarsza się rokowanie pacjentów [62].
Ostatnio okazało się, że klasyczne HN a także hartowanie na odległość mogą być przydatne w zapobieganiu uszkodzeniom okołoproceduralnym. Dla przykładu, w randomizowanej pracy obejmującej 150 pacjentów wykazano, że HN (dwie 90 sekundowe inflacje balona w odstępie 5 min) poprzedzające planowaną plastykę wieńcową skutkowało znamienną
redukcją występowania uszkodzenia okołoproceduralnego. W grupie kontrolnej i badanej
uszkodzenie to występowało, odpowiednio, u 25% i 7% pacjentów [63].
Ryc. 6.6. Dwa mechanizmy okołoproceduralnego uszkodzenia miokardium towarzyszącego planowanej plastyce wieńcowej. A. Przemieszczenie blaszki do ujścia odgałęzienia t. wieńcowej i niedrożność
tego odgałęzienia. B. Mikroembolizacja mikrokrążenia dystalnie do zwężenia.
144
Podobnie wykazano, że procedura hartowania na odległość (trzy 5-minutowe zaciskanie
tętnicy ramieniowej w 5-minutowych odstępach) wykonana tuż przed planowanym zabiegiem plastyki wieńcowej [64] lub przed operacją pomostowania tętnic wieńcowych znamiennie zmniejsza uwalnianie markerów martwicy miokardium [65,66], a w przypadku pomostowania, dodatkowo także częstość występowania ostrej niewydolności nerek [67].
Piśmiennictwo
1. Braunwald E, Maroko PR. The reduction of infarct size--an idea whose time (for testing) has come.
Circulation 1974; 50:206-209.
2. Reimer KA, Lowe JE, Rasmussen MM, Jennings RB. The wavefront phenomenon of ischemic
cell death. 1. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation 1977;
56:786-794.
3. Reimer KA, Jennings RB. The “wavefront phenomenon” of myocardial ischemic cell death. II. Transmural progression of necrosis within the framework of ischemic bed size (myocardium at risk) and
collateral flow. Lab Invest 1979; 40:633-644.
4. DeWood M, Spores J, Notske R, et al. Prevalence of total coronary occlusion during the early hours
of transmural myocardial infarction. N Engl J Med 1980; 303:897-902.
5. Downey JM, Cohen MV. Why do we still not have cardioprotective drugs? CIRC J 2009; 73:1171-1177.
6. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med 2007; 357:1121-1135.
7. Hausenloy DJ, Baxter G, Bell R et al. Translating novel strategies for cardioprotection: the Hatter
Workshop Recommendations. Basic Res Cardiol 2010; 105:677-686.
8. Levine HJ. Rest heart rate and life expectancy. J Am Coll Cardiol 1997; 30:1104-1106.
9. Beręsewicz A. Zwalnianie czynności serca poprzez blokowanie prądu rozrusznikowego If. Nowa
strategia terapeutyczna w kardiologii. Folia Cardiol 2005; 12 (Supl F):1-15.
10. Beręsewicz A, Kurzelewski M. Niestabilna blaszka miażdżycowa. Kard Pol 2001; 54:431-439.
11. Colin P, Ghaleh B, Monnet X, Hittinger L, Berdeaux A. Effect of graded heart rate reduction with
ivabradine on myocardial oxygen consumption and diastolic time in exercising dogs. J Pharmacol
Exp Ther 2004; 308:236-240.
12. Matsuzaki M, Patritti J, Tajimi T, Miller M, Kemper WS, Ross J, Jr. Effects of beta-blockade on regional myocardial flow and function during exercise. Am J Physiol 1984; 247:H52-H60.
13. Guth BD, Heusch G, Seitelberger R, Ross J, Jr. Mechanism of beneficial effect of beta-adrenergic
blockade on exercise-induced myocardial ischemia in conscious dogs. Circ Res 1987; 60:738-746.
14. Borer JS, Fox K, Jaillon P, Lerebours G. Antianginal and antiischemic effects of ivabradine, an I(f)
inhibitor, in stable angina: a randomized, double-blind, multicentered, placebo-controlled trial. Circulation 2003; 107:817-823.
15. Difrancesco D, Camm JA. Heart rate lowering by specific and selective If current inhibition with
ivabradine: a new therapeutic perspective in cardiovascular disease. Drugs 2004; 64:1757-1765.
16. Habib N. Reappraisal of heart rate as a risk factor in the general population. Eur Heart J Supplements
1999; 1 (Suppl H):H2-H10.
17. Palatini P. Elevated heart rate as a predictor of increased cardiovascular morbidity. J Hypertension
1999; 17 (Suppl. 3):S3-S10.
18. Borer JS. Drug insight: I(f) inhibitors as specific heart-rate-reducing agents. Nature Clinical Practice
Cardiovasc Med 2004; 1:103-109.
19. Bode-Boger SM, Kojda G. Organic nitrates in cardiovascular disease. Cell Mol Biol (Noisy-le-grand)
2005; 51:307-320.
145
20. Klemenska E, Beręsewicz A. Bioactivation of organic nitrates and the mechanism of nitrate tolerance. Cardiol J 2009; 16:11-19.
21. Mayer B, Beretta M. The enigma of nitroglycerin bioactivation and nitrate tolerance: news, views and
troubles. Br J Pharmacol 2008; 155:170-184.
22. Gori T, Parker JD. Nitrate-induced toxicity and preconditioning: a rationale for reconsidering the
use of these drugs. J Am Coll Cardiol 2008; 52:251-254.
23. Wenzel P, Mollnau H, Oelze M et al. First evidence for a crosstalk between mitochondrial and NADPH oxidase-derived reactive oxygen species in nitroglycerin-triggered vascular dysfunction. Antioxid Redox Signal 2008; 10:1435-1447.
24. Li Y, Zhang D, Jin W et al. Mitochondrial aldehyde dehydrogenase-2 (ALDH2) Glu504Lys polymorphism contributes to the variation in efficacy of sublingual nitroglycerin. J Clin Invest 2006;
116:506-511.
25. Larson HN, Zhou J, Chen Z, Stamler JS, Weiner H, Hurley TD. Structural and functional consequences of coenzyme binding to the inactive asian variant of mitochondrial aldehyde dehydrogenase: roles of residues 475 and 487. J Biol Chem 2007; 282:12940-12950.
26. Mackenzie IS, Maki-Petaja KM, Mceniery CM et al. Aldehyde dehydrogenase 2 plays a role in the
bioactivation of nitroglycerin in humans. Arterioscler Thromb Vasc Biol 2005; 25:1891-1895.
27. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in
health and disease. Physiol Rev 2010; 90:207-258.
28. Lam A, Lopaschuk GD. Anti-anginal effects of partial fatty acid oxidation inhibitors. Curr Opin
Pharmacol 2007; 7:179-185.
29. Bothe W, Olschewski M, Beyersdorf F, Doenst T. Glucose-insulin-potassium in cardiac surgery:
a meta-analysis. Ann Thorac Surg 2004; 78:1650-1657.
30. Timmer JR, Svilaas T, Ottervanger JP et al. Glucose-insulin-potassium infusion in patients with
acute myocardial infarction without signs of heart failure: the Glucose-Insulin-Potassium Study
(GIPS)-II. J Am Coll Cardiol 2006; 47:1730-1731.
31. van dH, I, Zijlstra F, van’t Hof AW et al. Glucose-insulin-potassium infusion inpatients treated with
primary angioplasty for acute myocardial infarction: the glucose-insulin-potassium study: a randomized trial. J Am Coll Cardiol 2003; 42:784-791.
32. Apstein CS. Glucose-insulin-potassium infusion and mortality in the CREATE-ECLA trial. JAMA
2005; 293:2596-2597.
33. Chaitman BR, Skettino SL, Parker JO et al. Anti-ischemic effects and long-term survival during ranolazine monotherapy in patients with chronic severe angina. J Am Coll Cardiol 2004; 43:1375-1382.
34. Stone PH, Gratsiansky NA, Blokhin A, Huang IZ, Meng L. Antianginal efficacy of ranolazine when
added to treatment with amlodipine: the ERICA (Efficacy of Ranolazine in Chronic Angina) trial. J
Am Coll Cardiol 2006; 48:566-575.
35. Mega JL, Hochman JS, Scirica BM et al. Clinical features and outcomes of women with unstable
ischemic heart disease: observations from metabolic efficiency with ranolazine for less ischemia in
non-ST-elevation acute coronary syndromes-thrombolysis in myocardial infarction 36 (MERLINTIMI 36). Circulation 2010; 121:1809-1817.
36. Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in
ischemic myocardium. Circulation 1986; 74:1124-1136.
37. Zhao ZQ, Corvera JS, Halkos ME et al. Inhibition of myocardial injury by ischemic postconditioning
during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol
2003; 285:H579-H588.
38. Kavazis AN. Exercise preconditioning of the myocardium. Sports Med 2009; 39:923-935.
146
39. Hausenloy DJ, Ong SB, Yellon DM. The mitochondrial permeability transition pore as a target for
preconditioning and postconditioning. Basic Res Cardiol 2009; 104:189-202.
40. Przyklenk K, Kloner RA. Ischemic preconditioning: exploring the paradox. Prog Cardiovasc Dis
1998; 40:517-547.
41. Beręsewicz A. Hartowanie niedokrwieniem – endogenny mechanizm kardioprotekcyjny. Annals of
Diagnostic Paediatric Pathology 2003; 7:37-43.
42. Vinten-Johansen J, Zhao ZQ, Jiang R, Zatta AJ, Dobson GP. Preconditioning and postconditioning:
innate cardioprotection from ischemia-reperfusion injury. J Appl Physiol 2007; 103:1441-1448.
43. Murphy E, Steenbergen C. Preconditioning: the mitochondrial connection. Annu Rev Physiol 2007;
69:51-67.
44. Downey JM, Davis AM, Cohen MV. Signaling pathways in ischemic preconditioning. Heart Fail Rev
2007; 12:181-188.
45. Burley DS, Ferdinandy P, Baxter GF. Cyclic GMP and protein kinase-G in myocardial ischaemiareperfusion: opportunities and obstacles for survival signaling. Br J Pharmacol 2007; 152:855-869.
46. Hausenloy DJ, Yellon DM. Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection. Heart Fail Rev 2007; 12:217-234.
47. Miura T, Tanno M, Sato T. Mitochondrial kinase signalling pathways in myocardial protection from
ischaemia/reperfusion-induced necrosis. Cardiovasc Res 2010; 88:7-15.
48. Bolli R. Preconditioning: a paradigm shift in the biology of myocardial ischemia. Am J Physiol Heart
Circ Physiol 2007; 292:H19-H27.
49. Jaffe MD, Quinn NK. Warm-up phenomenon in angina pectoris. Lancet 1980; 2:934-936.
50. Bogaty P, Kingma JG, Robitaille NM et al. Attenuation of myocardial ischemia with repeated exercise
in subjects with chronic stable angina - relation to myocardial contractility, intensity of exercise and
the adenosine triphosphate-sensitive potassium channel. J Am Coll Cardiol 1998; 32:1665-1671.
51. Tomai F. Warm up phenomenon and preconditioning in clinical practice. Heart 2002; 87:99-100.
52. Bilinska M, Rudnicki S, Beresewicz A. Delayed attenuation of myocardial ischemia with repeated
exercise in subjects with stable angina: a possible model for the second window of protection? Basic
Res Cardiol 2000; 95:418-423.
53. Kloner RA, Shook T, Przyklenk K et al. Previous angina alters in-hospital outcome in TIMI 4: a clinical correlate to preconditioning? Circulation 1995; 91:37-45.
54. Kloner RA, Shook T, Antman EM et al. Prospective temporal analysis of the onset of preinfarction
angina versus outcome: an ancillary study in TIMI-9B. Circulation 1998; 97:1042-1045.
55. Zhao ZQ. Postconditioning in reperfusion injury: a status report. Cardiovasc Drugs Ther 2010;
24:265-279.
56. Staat P, Rioufol G, Piot C et al. Postconditioning the human heart. Circulation 2005; 112:21432148.
57. Ma X, Zhang X, Li C, Luo M. Effect of postconditioning on coronary blood flow velocity and endothelial function and LV recovery after myocardial infarction. J Interv Cardiol 2006; 19:367-375.
58. Darling CE, Solari PB, Smith CS, Furman MI, Przyklenk K. ‘Postconditioning’ the human heart:
multiple balloon inflations during primary angioplasty may confer cardioprotection. Basic Res Cardiol 2007; 102:274-278.
59. Thibault H, Piot C, Staat P et al. Long-term benefit of postconditioning. Circulation 2008; 117:10371044.
60. Zhao WS, Xu L, Wang LF et al. A 60-s postconditioning protocol by percutaneous coronary intervention inhibits myocardial apoptosis in patients with acute myocardial infarction. Apoptosis 2009;
14:1204-1211.
147
61. Piot C, Croisille P, Staat P et al. M. Effect of cyclosporine on reperfusion injury in acute myocardial
infarction. N Engl J Med 2008; 359:473-481.
62. Babu GG, Walker JM, Yellon DM, Hausenloy DJ. Peri-procedural myocardial injury during percutaneous coronary intervention: an important target for cardioprotection. Eur Heart J 2011; 32:23-31.
63. Laskey WK. Beneficial impact of preconditioning during PTCA on creatine kinase release. Circulation 1999; 99:2085-2089.
64. Hoole SP, Heck PM, Sharples L et al. DP. Cardiac Remote Ischemic Preconditioning in Coronary Stenting (CRISP Stent) Study: a prospective, randomized control trial. Circulation 2009; 119:820-827.
65. Hausenloy DJ, Mwamure PK, Venugopal V et al. Effect of remote ischaemic preconditioning on
myocardial injury in patients undergoing coronary artery bypass graft surgery: a randomised controlled trial. Lancet 2007; 370:575-579.
66. Venugopal V, Hausenloy DJ, Ludman A et al. Remote ischaemic preconditioning reduces myocardial
injury in patients undergoing cardiac surgery with cold-blood cardioplegia: a randomised controlled
trial. Heart 2009; 95:1567-1571.
67. Venugopal V, Laing CM, Ludman A, Yellon DM, Hausenloy D. Effect of remote ischemic preconditioning on acute kidney injury in nondiabetic patients undergoing coronary artery bypass graft
surgery: a secondary analysis of 2 small randomized trials. Am J Kidney Dis 2010; 56:1043-1049.
148
VII. Patofizjologia miażdżycy
i ostrych zespołów wieńcowych
Andrzej BERĘSEWICZ, Emilia KLEMENSKA
Miażdżyca jest „chorobą zapalną” w dwóch znaczeniach [1]:
1. Jest to zlokalizowany proces zapalny toczący się w ścianie dużych i średnich tętnic i skutkujący
powstawaniem typowych ogniskowych zmian naczyniowych. Początkowo są to pasma tłuszczowe (fatty streaks), będące skupiskami komórek zapalnych (głównie przekształconych makrofagów) w intimie. Z czasem, w wyniku interakcji procesów zapalnego i reparacyjnego, powstają
zaawansowane zmiany naczyniowe, znane jako blaszki miażdżycowe (atherosclerotic plaque)
(ryc. 1.3.). Dopiero one są źródłem ewentualnych objawów klinicznych (rozdz. VII.2);
2. Miażdżycy towarzyszy często ogólnoustrojowy podostry odczyn zapalny (systemic lowgrade inflammation). Jego wykładnikami są zwiększone poziomy we krwi: (i) cytokin prozapalnych (np. TNF-α, interleukina-6); (ii) rozpuszczalnych białek adhezyjnych (ICAM,
VCAM, selektyny) oraz (iii) tzw. białek ostrej fazy zapalenia (np. CRP, amyloid A, fibrynogen). Badania kliniczne dowodzą, że poziomy tych markerów są czynnikami ryzyka wydarzeń sercowo-naczyniowych [12]. Stąd hipoteza, że systemowy odczyn zapalny bierze
udział w mechanizmie destabilizacji już uformowanych blaszek miażdżycowych [13], która
to destabilizacja jest elementem mechanizmu OZW (rozdz. VII.4.2.1).
Od niedawna wiadomo, że zapalenie towarzyszące miażdżycy ma taki sam mechanizm
jak „klasyczne” zapalenie towarzyszące infekcjom, i że oba te zapalenia są wynikiem aktywacji
wrodzonego systemu odporności (innate immunity) [2-11]. Główna różnica polega na tym,
że w infekcjach, induktorem wrodzonej odporności i zapalenia jest patogen lub jego toksyna,
a w miażdżycy induktorem są lipoproteiny o małej gęstości, które uległy modyfikacji chemicznej pod wpływem ROS (oksydowane LDL, ox-LDL).
Mechanizm i sens biologiczny zapalenia, niezależnie od mechanizmu aktywacji, sprowadzają się do następujących punktów:
1. W ścianie naczyń i w innych tkankach stale rezydują komórki wrodzonej odporności, w tym głównie makrofagi. W kontakcie w czynnikami aktywującymi (np. bakterie,
149
ox-LDL) komórki te produkują substancje, znane jako mediatory zapalenia (w tym chemokiny i cytokiny). Mediatory promują rekrutację dodatkowych komórek zapalnych z krążenia do ogniska zapalenia. Są także aktywatorami procesów naprawczych;
2. Powstały w ten sposób odczyn zapalny jest reakcją ochronną mającą na celu: (i) unieszkodliwienie i separację od otoczenia czynnika uszkadzającego i uszkodzonej tkanki oraz (ii)
gojenie i reparację uszkodzenia;
3. Zapalenie toczy się zazwyczaj lokalnie w naczyniach krwionośnych i otaczającej tkance, ale
poprzez mediatory zapalenia może mieć oddziaływania ogólnoustrojowe;
4. Zapalenie ustępuje z chwilą eliminacji czynnika inicjującego, dlatego, w zasadzie, jest procesem samoograniczającym się. Powodem przejścia zapalenia w fazę przewlekłą jest stała
obecność czynnika wyzwalającego zapalenie. W fazie przewlekłej, obraz choroby może być
zdominowany przez niekorzystne efekty zapalenia (vide miażdżyca).
W kolejnych częściach tego rozdziału omawiamy:
1. mechanizm „klasycznego” systemu odporności wrodzonej oraz receptory tego systemu,
o których już dzisiaj wiadomo, że biorą udział w procesie miażdżycowym, i aktywacja których jest prawdopodobną przyczyną systemowego podostrego stanu zapalnego towarzyszącego otyłości i cukrzycy (rozdz. VII.1);
2. mechanizm, w jakim w ścianie naczyniowej powstają ox-LDL, i w jakim, poprzez aktywację
odporności wrodzonej, ox-LDL skutkują powstaniem ogniskowych zmian miażdżycowych
(rozdz. VII.2). Prawdopodobnym źródłem ox-LDL jest naczyniowy stres oksydacyjny,
o którym wiadomo, że jest procesem dotyczącym całego drzewa naczyniowego. Dlatego
niejasny jest powód, dla którego zmiany miażdżycowe mają charakter ogniskowy. Także
mechanizm naczyniowego stresu oksydacyjnego jest tylko częściowo poznany;
3. konsekwencje kliniczne miażdżycowych zmian naczyniowych (rozdz. VII.3); oraz
4. mechanizm OZW, o którym wiadomo, że ma związek z przetrwałą aktywnością i/lub ponowną aktywacją zapalenia w niektórych dojrzałych blaszkach miażdżycowych (rozdz.
VII.4). Przez lata sądzono, że zwiększone poziomy markerów zapalenia, obserwowane
u pacjentów z miażdżycą, są zjawiskiem towarzyszącym – biernym markerem lokalnego
zapalenia w blaszkach. Obecnie panuje opinia, że systemowy podostry odczyn zapalny jest
raczej przyczyną, a nie skutkiem, lokalnej aktywacji zapalenia w blaszkach i ich destabilizacji [12,13]. W tym kontekście omawiamy kilka aktualnych hipotez tłumaczących mechanizm uogólnionego odczynu zapalnego w miażdżycy (rozdz. VII.4.2.4).
VII.1. Wrodzona odporność i jej udział w mechanizmie miażdżycy
VII.1.1. Repetytorium z mechanizmów wrodzonej odporności
System wrodzonej odporności składa się z komórek i mechanizmów, które w sposób niespecyficzny chronią organizm gospodarza przed zakażeniem przez inne organizmy. Komórki
wrodzonej odporności rozpoznają patogeny i reagują na nie w oparciu o identyfikację jedynie
generalnych różnic gatunkowych między patogenem i gospodarzem (rozróżnianie na zasadzie
„swój-obcy”). W przeciwieństwie do młodszego ewolucyjnie systemu nabytej odporności, system odporności wrodzonej: (i) umożliwia prawie natychmiastową odpowiedź obronną na zakażenie, ale (ii) jest to odpowiedź całkowicie nieswoista i (iii) krótkotrwała, gdyż ustępuje z chwilą
150
eliminacji patogenu i brak jest mechanizmu pamięci skutkującego powstaniem długotrwałej
zapobiegawczej odporności. Mechanizm wrodzonej odporności sumuje ryc. 7.1.
Ryc. 7.1. Uproszczony schemat mechanizmu, w jakim aktywacja wrodzonej odporności skutkuje lokalnym i systemowym odczynem zapalnym. SR, receptory zmiatające; TLR, receptory Toll-podobne.
Szczegóły w tekście.
Wrodzoną odporność, skutkującą zapaleniem i fagocytozą, wyzwala aktywacja, tzw. receptorów PRR (pattern recognition receptors) przez odpowiednie ligandy (vide poniżej). Receptory te, pełniące rolę systemu „wczesnego ostrzegania” są obecne głównie na rezydujących
w tkankach makrofagach, komórkach dendrytycznych i komórkach NK. Ostatnio okazało
się, że są obecne także na różnych niezapalnych komórkach, w tym na komórkach śródbłonka i naczyniowych mięśni gładkich. Istnieje kilka klas receptorów PRR. Z punktu widzenia
patofizjologii układu sercowo-naczyniowego i miażdżycy szczególnie ważne są: (i) receptory zmiatające (SR, scavenger receptors) [2] (rozdz. VII.1.1) oraz (ii) receptory Toll-podobne
(TLR, toll-like receptors, podobne do białka Toll scharakteryzowanego pierwotnie u muszki
owocowej, ang. Toll oznacza cło, komorę celną) (rozdz. VII.1.2.) [7,8,11].
Pierwotnie przypuszczano, że ligandami PRR i czynnikami uruchamiającymi mechanizm
wrodzonej odporności są silnie konserwatywne i wspólne dla różnych gatunków patogenów
ugrupowania cząsteczkowe (epitopy). Klasycznym przykładem takiego czynnika jest lipopolisacharyd błon komórkowych bakterii Gram ujemnych (LPS). Czynniki te opatrzono akronimem
PAMPs (Pathogen Associated Molecular Patterns) gdyż pierwotnie zakładano, że wrodzony system odpornościowy rozpoznaje wzory cząsteczkowe wyłącznie wg zasady „swój-obcy”. Dzisiaj
wiadomo, że ligandami PRR są różne endogenne struktury/cząsteczki, których powstawanie ma
151
związek z działaniem czynników uszkadzających, w tym ROS, na tkanki gospodarza (ryc. 7.2.).
Dla odróżnienia, te endogenne ligandy PRR otrzymały akronim DAMPs (Damage Associated
Molecular Patterns). PAMPs i DAMPs noszą wspólną nazwę alarminy.
Fakt, że PAMPS i DAMPs są ligandami tych samych receptorów sugeruje, że ich struktura
molekularna jest podobna. Istnieje także możliwość, że ewolucja rozwinęła wrażliwość na
DAMPs, dlatego że odczyn zapalny i uszkodzenie tkanek zwykle współistnieją z sobą i fakt, że
produkty molekularne uszkodzenia są czynnikami sygnałowymi ułatwia aktywację procesów
reparacyjnych. Podobne rozumowanie dotyczy faktu, że miażdżyco-rodne ox-LDL są rozpoznawane przez receptory zmiatające i TLR (ryc. 7.2.). Otóż odczynowi zapalnemu, aktywacji
komórek zapalnych i apoptozie z reguły towarzyszy zwiększona produkcja ROS, co prawdopodobnie skutkuje dość standardową wolnorodnikową modyfikacją różnych struktur fosfolipidowych i białkowych, zarówno w obrębie komórek jak i LDL. Liczne dane eksperymentalne
pokazują, że DAPMs powstające w wyniku utleniania (oxidation-specific epitopes) są rozpoznawane przez receptory PRR, aktywują system wrodzonej odporności i prawdopodobnie są
odpowiedzialne za rozwój procesu miażdżycowego [14].
Ryc. 7.2. Egzogenne i endogenne ligandy receptorów PRR odpowiedzialnych za aktywację wrodzonej
odporności. Ligandami niektórych PRR są, obok ox-LDL, nasycone kwasy tłuszczowe oraz zaawansowane produkty glikacji białek (AGE), co tłumaczy związek zapalenia z otyłością oraz cukrzycą. ECM,
macierz pozakomórkowa.
Ligand związany z receptorem wymiatającym (bakterie, komórki apoptotyczne, aktywowane płytki krwi czy ox-LDL) podlega fagocytozie a następnie degradacji w lizosomach.
Równocześnie, aktywacja tych receptorów skutkuje słabo poznaną kaskadą sygnałów komórkowych (prawdopodobnie różniącą się między poszczególnymi klasami receptorów), ale
ostatecznie skutkującą aktywacją rodziny czynników transkrypcyjnych NF-кB (nuclear factor
kappa B) i zwiększoną ekspresją tzw. genów i białek zapalenia (ryc. 6.1.) (vide poniżej).
152
Podobnie, aktywacja receptorów TLR uruchamia wewnątrzkomórkowe ścieżki sygnalizacyjne, które generalnie kończą się (i) aktywacją NF-кB i produkcją pro-zapalnych białek oraz (ii) aktywacją czynnika transkrypcyjnego IRF (interferon regulatory factor) i produkcją interferonów α i β.
Jak widać, NF-кB jest wspólnym efektorem aktywacji receptorów TLR i zmiatających. Wiadomo
także, że obok aktywacji receptorów PRR, różne inne czynniki skutkują aktywacją NF-кB, w tym:
(i) cytokiny pro-zapalne TNF-α, IL-6 i inne; (ii) stres oksydacyjny oraz (iii) niektóre hormony
i czynniki wzrostowe (np. endotelina-1, insulina, PAF, trombosan, IGF-1, EGF, TGF-α) [15].
W układach eksperymentalnych wykazano, że aktywacja NF-кB skutkuje zwiększoną
ekspresją kilkudziesięciu genów i białek. Nie jest jednak pewne czy w warunkach in vivo
wszystkie te geny są aktywowane równocześnie, równie silnie i podobnie w różnych rodzajach
komórek. W kontekście patomechanizmu miażdżycy prawdopodobnie najważniejsze białka
zależne od NF-кB to (vide www.nf-kb.org):
1. Pro-zapalne chemokiny i ich receptory, czyli białka o działaniu chemotaktycznym w stosunku do makrofagów i innych komórek. Przykładem takiej chemokiny jest białko MCP-1
(monocyte chemotactic/chemoattractant protein 1);
2. Czynniki wzrostowe i ich receptory, w tym odpowiedzialne za transformację leukocytów
G-CSF (granulocyte colony stimulating factor) i GM-CSF (granulocyte macrophage colony
stimulatinf factor);
3. Zapalne cytokiny i ich receptory, czyli generalnie białka o działaniu aktywującym komórki zapalne i zwiększającym ich zdolność do eliminacji patogenów między innymi w mechanizmie
apoptozy, w tym interleukiny IL-6, IL-1β i czynnik martwicy nowotworów α (TNF-α);
4. Niektóre receptory PRR, w tym TLR-2 i TRL-9, receptor wymiatający LOX-1 (rozdz.
VII.1.1.), receptor wymiatający AGE (RAGE);
5. Cząsteczki adhezyjne, w tym obecne na komórkach śródbłonka P-selektyna, E-selektyna,
ICAM-1 i VCAM-1;
6. Różne czynniki o działaniu pro-apoptotycznym, w tym tzw. receptor śmierci Fas (pro-apoptotic receptor) i jego leukocytarny ligand FAS-L (FAS ligand) (ryc. 5.13.);
7. Enzymy, w tym indukowana izoforma syntazy NO (iNOS), cyklooksygenaza-2 (COX-2),
fosfolipaza A2 (PLA2, vide ryc. V-10), dyzmutaza ponadtlenkowa (SOD, ryc. 8.1.) czy oksydaza NADPH (enzym odpowiedzialny za produkcję ROS przez komórki fagocytujące, ale
także przez komórki ściany naczyniowej (ryc. 8.3.);
8. Enzymy trawiące białka macierzy pozakomórkowej, w tym metaloproteinazy 1, 3 i 9;
9. Białka biorące udział w prezentacji antygenu.
Opisana powyżej kaskada wrodzonej odporności i odczynu zapalnego ma na celu:
1. Rekrutację, przy pomocy chemicznych mediatorów, komórek odpornościowych do miejsca infekcji, czy uszkodzenia tkanki;
2. Aktywację mechanizmów nastawionych na unieszkodliwianie a następnie usuwanie nieżywych patogenów, uszkodzonych fragmentów tkanki oraz obcych lub zmodyfikowanych
substancji;
3. Udział w procesach reparacyjnych;
4. Aktywację nabytego systemu odporności w mechanizmie prezentacji antygenów.
Kaskada wrodzonej odporności i odczynu zapalnego posiada liczne mechanizmy wzmacniające. Dla przykładu:
153
1. Mediatory odczynu zapalnego, jakimi są cytokiny (np. IL-6, TNF-α) są silnymi aktywatorami NF-кB i odczynu zapalnego;
2. Lokalny odczyn zapalny może skutkować wzrostem poziomu cytokin pro-zapalnych we
krwi (TNF-α, IL-6) i wtedy stają się one bodźcem dla wątroby do zwiększonej produkcji
kilkudziesięciu tzw. białek ostrej fazy zapalenia, w tym CRP, fibrynogenu, amyloidu-A i angiotensynogenu. Działanie takich substancji jak CRP i amyloid A jest tylko częściowo poznane (rozdz. VII.4.2.4), ale dzięki temu, że są one ligandami receptorów PRR ich działanie
polega, między innymi, na wzmacnianiu pierwotnego odczynu zapalnego (ryc. 7.1., lewa
strona). CRP jest ligandem receptorów zmiatających LOX-1 i SR-A [16-18] a amyloid A
ligandem receptorów zmiatających i receptorów TLR-2 [3].
3. Lokalny odczyn zapalny obok działania unieszkodliwiającego patogeny, może mieć działanie uszkadzające na otaczające tkanki między innymi poprzez indukowanie stresu oksydacyjnego i apoptozy. W ten sposób lokalny odczyn zapalny może być źródłem DAMPs, które
wydostając się poza pierwotne ognisko zakażenia/uszkodzenia podtrzymują i/lub wzmacniają pierwotny czynnik pozapalny (ryc. 7.1., prawa strona). Pod wpływem DAMPs dochodzi do indukcji „aseptycznej” kaskady stanu zapalnego, której sens biologiczny polega
na usuwaniu uszkodzonych elementów tkanek (np. aseptyczny odczyn zapalny w ognisku
niedokrwienia/reperfuzji [19], usuwanie zmodyfikowanych LDL, etc.) a następnie promocji procesów gojenia się tkanki (np. bliznowacenie) i ewentualnie procesów naprawczych
(np. rekrutacja komórek macierzystych).
Kaskada reakcji składających się na wrodzoną odporność ma, w zasadzie, przebieg samoograniczający się i wygasający wraz z eliminacją PAMPs/DAMPs (vide zapalenie w obszarze
niedokrwionego/reperfundowanego miokardium). Niemniej jednak, coraz więcej obserwacji dowodzi udziału mechanizmów wrodzonej odporności w różnych stanach chorobowych,
które, jak miażdżyca czy reumatyczne zapalenie stawów [10], są przewlekłymi chorobami
zapalnymi, bądź którym towarzyszy podostre systemowe zapalenie. W przypadku stanów
kardiologicznych dotyczy to głównie niewydolności serca [20], oraz stanów składających się
na zespół kardio-metaboliczny [9,71] (rozdz. X). Przewlekła ekspozycja ustroju na PAMPs/
DAMPs może być jedną z przyczyn przechodzenia odczynu zapalnego w stadium przewlekłe,
ale generalnie mechanizm tego procesu jest słabo zrozumiały.
VII.1.2. Receptory zmiatające biorące udział w powstawaniu miażdżycy
Zidentyfikowano dotąd kilkanaście receptorów zmiatających (SR), które ze względu na
budowę, zostały podzielone na 8 klas. Nie jest jasne czy ta wielość receptorów ma służyć rozpoznawaniu różnych patogenów, czy może generowaniu różnych odpowiedzi biologicznych.
W każdym razie, o pięciu klasach tych receptorów (SR-A, SR-B, SR-E, SR-F i SR-G) wiadomo,
że ich ligandami są, między innymi, miażdżycogenne ox-LDL. W oparciu o badania, w których wykazano, że eliminacja genu danego receptora zapobiega, a jego nadekspresja wzmaga
eksperymentalną miażdżycę u myszy transgenicznych, wydaje się, że szczególną rolę w procesie powstawania zmian miażdżycowych odgrywają receptory CD36 (FAT/CD36) (przedstawiciel klasy SR-B) oraz LOX-1 (lectin-like ox-LDL receptor, przedstawiciel klasy SR-E)
[2,21]. Nowe dane sugerują, że obok działania lokalnego w miejscu powstawania zmian miażdżycowych w naczyniach, receptory CD36 i LOX-1 pełnią rolę „sensora” stanu metaboliczne154
go ustroju i w tej roli są induktorem systemowego zapalenia i insulinooporności, jakie towarzyszą otyłości, cukrzycy typu II, czy zespołowi metabolicznemu [9,22].
Receptory zmiatające CD36 (przedstawiciel klasy SR-B); Są to wielofunkcyjne glikoproteiny błonowe, które równocześnie pełnią rolę: (i) receptora wymiatającego; (ii) koaktywatora receptorów TLR 2 i 6 oraz (iii) dokomórkowego transportera niezestryfikowanych kwasów tłuszczowych (FAT/CD36, vide rozdz. IV.4). Poza monocytami/makrofagami, CD36 występuje także
na wielu innych komórkach, w tym komórkach naczyniowych i komórkach tłuszczowych.
Receptory CD36 rozpoznają różne ligandy, w tym ox-LDL, niezestryfikowane kwasy tłuszczowe (NEFA), AGE, powierzchnie komórek apoptotycznych, powierzchnie bakteryjne i inne.
Ustalono, że CD36 rozpoznają w obrębie ox-LDL utlenione cząsteczki fosfatydylcholiny, jednego
z fosfolipidów budujących LDL. Wykazano ponadto, że cząsteczki utlenionej fosfatydylcholiny są ligandem, za pomocą którego CD36 rozpoznaje komórki apoptotyczne i bakterie [2,14]. W układach
eksperymentalnych wykazano, że CD36 jest głównym receptorem odpowiedzialnym za wiązanie
i internalizację ox-LDL przez makrofagi. Zwiększoną gęstość CD36 wykazano w naczyniowych
zmianach miażdżycowych. U myszy pozbawionych genu CD36, rozwój zmian miażdżycowych jest
bardzo ograniczony, co razem wziąwszy dowodzi roli CD36 w procesie aterogenezy [2,9].
Wiele faktów sugeruje, że obok miażdżycy, CD36 mogą promować występowanie systemowego podostrego nieinfekcyjnego zapalenia, jakie zwykle towarzyszy otyłości, cukrzycy
typy II i ogólnie insulinooporności i zespołowi metabolicznemu (ryc. 7.3.).
Ryc. 7.3. Model mechanizmu, w którym interakcja zaburzeń metabolicznych z FAT/CD36 skutkuje powstawaniem zmian miażdżycowych, systemowym stanem zapalnym i insulinoopornością. Zespołowi metabolicznemu i jego komponentom towarzyszy wzrost ekspresji CD36 [23]. Zwiększone poziomy lipidów,
działając jako DAPMs, aktywują wrodzoną odporność, co skutkuje lokalnym gromadzeniem komórek
piankowatych, systemowym wzrostem poziomu markerów zapalenia oraz fosforylacją IRS-1 (poprzez aktywację JNK). Fosforylacja IRS-1 skutkuje mniejszą aktywnością kinazy Akt w różnych tkankach, w tym
w mięśniach i tkance tłuszczowej, mniejszą ekspresją Glut-4 na powierzchni komórek i zaburzeniami metabolizmu glukozy. IRS-1, insulin receptor substrate-1; JNK, c-Jun N-terminal kinase. Wg [9,24].
155
Przemawiają za tym następujące fakty (przegląd literatury w ref. [9]):
1. Takie pro-miażdżycowe czynniki jak ox-LDL, hiperglikemia, hiperinsulinemia, otyłość,
cukrzyca typu II zwiększają ekspresję CD36 w makrofagach i innych tkankach [9];
2. Ligandem CD36 indukującym reakcję wrodzonej odporności są między innymi długołańcuchowe nienasycone kwasy tłuszczowe (NEFA), których poziomy są zwiększone w stanach
chorobowych składających się na zespół metaboliczny. Nawiasem mówiąc NEFA aktywują
CD36 wtedy, kiedy pełni on rolę koaktywatora TLR 2 i 4;
3. Dieta bogatotłuszczowa indukuje systemowy podostry stan zapalny u zwierząt eksperymentalnych, którego objawami są: zwiększone poziomy we krwi zapalnych chemokin i cytokin i zwiększona infiltracja makrofagów w tkance tłuszczowej. Wykazano, że eliminacja
genu CD36 zapobiega temu odczynowi;
4. Redukcja ekspresji CD36 zapobiegała rozwojowi insulinooporności u szczepu myszek
z wrodzoną otyłością (ob/ob);
5. Eliminacja w makrofagach genu odpowiedzialnego za produkcję MCP-1 zapobiegała rozwojowi insulinooporności u zwierząt karmionych dietą bogatotłuszczową;
6. Badania in vitro wykazały, że ox-LDL, działając poprzez aktywację CD36, powoduje w komórkach tłuszczowych aktywację kaskady MAP-kinaz (konkretnie kinazy INK), co skutkuje fosforylacją IRS-1 (insulin receptor substrate-1), upośledzeniem trandukcji sygnałów
przez receptor insulinowy i insulinoopornością komórek (ryc. 7.3.);
7. U pacjentów z cukrzycą, poziom ekspresji CD36 na monocytach/makrofagach, poziom
rozpuszczalnej postaci CD36 we krwi oraz poziom ox-LDL we krwi korelował z poziomem
glikemii (gorszej kontroli glikemii towarzyszyła większa ekspresja CD36).
Receptory LOX-1 (lectin-like ox-LDL receptor, przedstawiciel klasy SR-E) [22,25]. Obok
tego, że biorą udział w powstawaniu eksperymentalnej miażdżycy, o receptorach LOX-1 wiadomo, że:
1. Są konstytutywnie obecne na monocytach/makrofagach, komórkach śródbłonka, komórkach mięśni gładkich i płytkach krwi;
2. Wiążą a następnie inicjują fagocytozę różnych zmodyfikowanych chemicznie substancji i uszkodzonych komórek (ox-LDL, apoptotyczne/stare komórki, zaktywowane płytki,
bakterie);
3. Związane z ligandem, uruchamiają ścieżki sygnalizacyjne typowe dla wrodzonej odporności i odczynu zapalnego (rozdz. VII.1) (ryc. 7.4.);
4. ekspresja LOX-1 rośnie pod wpływem ox-LDL, angiotensyny, endoteliny, ROS, cytokin
pro-zapalnych, CRP i innych;
5. obok ox-LDL, także białko ostrej fazy CRP jest ligandem i czynnikiem zwiększającym
ekspresję LOX-1 [16] i, co prawdopodobnie ważne, komórkowe efekty CRP są bliźniaczo
podobne do efektów ox-LDL [12];
6. Blokery receptorów angiotensynowych AT1, statyny i kwas acetylosalicylowy, leki o których wiadomo, że zmniejszają ryzyko sercowo-naczyniowe, zmniejszały ekspresję LOX-1
w różnych badaniach in vitro;
7. Aktywacja LOX-1 skutkuje zwiększoną ekspresją receptorów angiotensynowych AT1 na
komórkach mięśni gładkich i makrofagach (element sprzężenia zwrotnego gdyż aktywacja AT1 ma działanie pro-zapalne i pro miażdżycowe);
156
8. U ludzi, zwiększoną ekspresję LOX-1 wykazano w naczyniach zmienionych miażdżycowo
a także w hipercholesterolemii, cukrzycy i nadciśnieniu;
9. rozpuszczalna postać LOX-1 jest obecna w surowicy w okresie poprzedzającym OZW,
więc jej pomiar może się okazać przydatny diagnostycznie;
10. polimorfizm genu LOX-1 jest częstszy u osób z zawałem w porównaniu z grupą kontrolną [22];
11. U pacjentów z hipercholesterolemią stwierdzono zwiększoną ekspresję LOX-1 na płytkach
krwi i ekspresja ta malała już po 9 dniach leczenia atorwastatyną (20 mg/dobę) [26].
Ryc. 7.4. Efekty interakcji ox-LDL z komórkami śródbłonka, komórkami mięśni gładkich i makrofagami. Ox-LDL, za pośrednictwem receptora LOX-1: (a) są w niekontrolowany sposób fagocytowane przez
makrofagi; (ii) aktywują komórki śródbłonka oraz indukują ekspresję substancji chemotaktycznych
w stosunku do komórek zapalnych i wobec tego są elementem sprzężenia zwrotnego podtrzymującego
proces aktywacji śródbłonka; (iii) aktywują makrofagi, które produkują zwiększone ilości substancji
chemotaktycznych, prozapalnych, czynników wzrostowych, ROS, pro-krzepliwego czynnika tkankowego i metaloproteinę (enzymy wyspecjalizowane w trawieniu elementów tkanki łączne). Liczne czynniki
zwiększają ekspresję LOX-1 (lewa strona). Zwiększona ekspresja LOX-1 towarzyszy czynnikom ryzyka
sercowo-naczyniowego u ludzi (prawa strona). MCP-1 – monocyte chemoattractant protein-1; M-CSF –
monocyte colony stimulating factor; VCAM-1 – vascular cell adhesion molekule-1 [22,25].
VII.1.3. Receptory TLR
Zidentyfikowano 10 rodzajów TRL u ludzi i 13 u myszy [7,8,11]. Badania eksperymentalne z udziałem transgenicznych myszy (eliminacja lub nadekspresja genu odpowiedniego
TLR) sugerują szczególną rolę receptorów TLR 2 i TLR 4 (obecne także u człowieka) i indukowanego przez nie procesu wrodzonej odporności w procesie rozwoju eksperymentalnej
miażdżycy [3,5]. Dodatkowe argumenty na rzecz tej hipotezy są następujące [5,10]:
1. Ligandami TLR-2 i 4 są bardzo liczne substancje, w tym ox-LDL, nasycone kwasy tłuszczowe, białko ostrej fazy amyloid A, różne składniki macierzy pozakomórkowej lub produkty
jej degradacji (fibronektyna, fibrynogen, biglikan, versikan, fragmenty siarczanu heparyny,
fragmenty kwasu hialuronowego), białka szoku cieplnego i inne [4];
157
2. TLR-2 i 4 są konstytutywnie obecne w naczyniach tętniczych i są praktycznie nieobecne
w żyłach. W zdrowych tętnicach wykrywa się je jedynie na komórkach dendrytycznych
rezydujących w intimie. Natomiast w ludzkich tętnicach objętych zmianą miażdżycową
stwierdza się zwiększoną ekspresję TLR-2 i 4 na komórkach śródbłonka, makrofagach, komórkach mięśni gładkich, fibroblastach i w mniejszym stopniu na limfocytach T;
3. Pacjenci z chorobą wieńcową mają zwiększoną ekspresję TLR-2 i 4 na monocytach izolowanych z krwi krążącej a u pacjentów z ostrym zespołem wieńcowym poziom tej ekspresji
koreluje z poziomem we krwi standardowych markerów zapalenia;
4. W zmianach miażdżycowych u ludzi wykazano ścisłą przestrzenną ko-lokalizację TLR-2
i 4 i aktywacji NF-кB;
5. Wykazano, że aktywacja TLR-2 jest głównym czynnikiem odpowiedzialnym za zwiększoną
produkcję MCP-1, IL-6 i metaloproteinaz MMP-1, -2, –3 i –9 w blaszkach miażdżycowych
ludzi (endartectomia tętnic szyjnych) [27].
VII.2. Mechanizm komórkowy ogniskowych naczyniowych zmian
miażdżycowych
Prawdopodobną sekwencję procesów pro-miażdżycowych ilustruje ryc. 7.5. Można jednak przypuszczać, że ten sekwencyjny schemat mechanizmu jest w wielu miejscach dodatkowo komplikowany przez pętle sprzężeń zwrotnych, które są elementem podtrzymującym
i/lub wzmacniającym pierwotny mechanizm miażdżycowy: [13,28-35].
Ryc. 7.5. Prawdopodobna sekwencja procesów za pośrednictwem, których tzw. czynniki ryzyka prowadzą do rozwoju ogniskowych naczyniowych zmian miażdżycowych i wydarzeń sercowo-naczyniowych.
158
1. Zgodnie z aktualną hipotezą, proces jest wyzwalany przez czynniki ryzyka choroby sercowo-naczyniowej;
2. W wyniku ich działania, w ścianie tętnic rośnie produkcja toksycznego anionorodnika ponadtlenkowego (O2–) i innych ROS. Źródłem O2– są z jednej strony naczyniowe enzymy:
oksydaza NADPH, oksydaza ksantynowa i ewentualnie śródbłonkowa syntaza NO, a z drugiej – oksydaza NADPH leukocytów „zadherowanych” do dysfunkcjonalnego śródbłonka
i/lub zgromadzonych w infimie (rozdz. VIII);
3. Efektem nadprodukcji O2– jest:
a. mniejsza biodostępność i/lub produkcja tlenku azotu (NO) w naczyniach, co skutkuje
dysfunkcją śródbłonka naczyniowego (rozdz. IX) oraz
b. oksydacyjna modyfikacja LDL, głównie tych które przechodzą przez barierę śródbłonkową w drodze do komórek docelowych (przechodzenie w ilości proporcjonalnej do
stężenia LDL we krwi) i w efekcie powstają różne postaci ox-LDL, które stają się ligandami (DAMPs) receptorów wrodzonego układu odpornościowego (zmiatających i TLR)
(rozdz. VII.1.2);
c. Nadmiar O2– i zmniejszona biodostępność NO (skutkujące dysfunkcją śródbłonkową)
oraz aktywacja przez ox-LDL receptorów wrodzonego systemu odpornościowego (ryc. 7.3.
i 7.4.) powodują tzw. aktywację komórek śródbłonka, której konsekwencją są: (i) ekspresja
różnych cząstek adhezyjnych na powierzchni śródbłonka. Z badań laboratoryjnych wiadomo, że eliminacja genu, przede wszystkim VCAM-1 i w mniejszym stopniu ICAM-1,
zapobiega powstawaniu eksperymentalnej miażdżycy; (ii) śródbłonkowa produkcja różnych substancji chemotaktycznych i cytokin pro-zapalnych i ich receptorów. Szczególnie
ważna jest produkcja chemokiny MCP-1 (monocyte chemotactic protein-1) gdyż eliminacja
jej genu (lub genu jej receptora) zapobiega rozwojowi eksperymentalnej miażdżycy. Obok
śródbłonka, źródłem MPC-1 są także komórki mięśni gładkich naczyń, kiedy są poddane
działaniu cytokin pozapalnych; (iii) destrukcja glikokaliksu śródbłonkowego oraz (iv) „rozszczelnienie” bariery śródbłonkowej co ułatwia przechodzenie przez śródbłonek LDL i monocytów oraz (iv) zmiana ekspresji licznych czynników śródbłonkowych wpływających na
proces krzepnięcia i fibrynolizy (tab. 7.2.), co razem wziąwszy jest wyrazem powstania tzw.
pro-zapalnego i pro-krzepliwego fenotypu śródbłonka (rozdz. IX).
4. Komórki śródbłonka, które normalnie przeciwdziałają adhezji płytek i komórek zapalnych
(rozdz. IX), poprzez zwiększoną ekspresję cząstek adhezyjnych, promują teraz adhezję
płytek i komórek zapalnych, głównie monocytów i limfocytów T. Komórki te, unieruchomione na powierzchni śródbłonka, przechodzą (pod wpływem chemoatraktantów) pod
śródbłonek, gdzie monocyty: (i) różnicują się do makrofagów; (ii) produkują liczne substancje czynne promujące dalszą ich rekrutację do intimy (iii) wiążą i fagocytują ox-LDL
i w ten sposób stają się komórkami piankowatymi oraz (iv) już jako komórki piankowate,
mnożą się lokalnie w infimie. Prawdopodobnymi czynnikami stymulującymi te podziały
makrofagów są cytokiny M-CSF (macrophage colony-stimulating factor) i GM-CSF (granulocyte-macrophage colony-stimulating factor), o których wiadomo, że są obecne w zmianach
miażdżycowych u ludzi, i że ich eliminacja opóźnia rozwój eksperymentalnej miażdżycy.
Makrofagi obładowane lipidami (a z czasem także komórki śródbłonka i naczyniowych
mięśni gładkich oraz fibroblasty fagocytujące ox-LDL przy udziale receptorów zmiatają159
cych) mają charakterystyczny wygląd, opisywany przez patologów jako komórki piankowate. Ich akumulacja w ścianie naczyniowej jest widoczna w postaci pasm tłuszczowych
i traktowana jako wczesna zmiana miażdżycowa. Komórki piankowate obumierają (m.in.
w wyniku apoptozy), a ich tłuszczowa zawartość wylewa się do przestrzeni pozakomórkowej i tworzy półpłynne jądro lipidowe blaszki miażdżycowej;
5. Uszkodzony śródbłonek, makrofagi, płytki krwi i komórki mięśni gładkich produkują, między
innymi, liczne czynniki wzrostowe, które uruchamiają procesy reparacyjne. Pod ich wpływem,
komórki mięśni gładkich ściany naczyniowej migrują z warstwy mięśniowej do ogniska miażdżycowego. Tam mnożąc się, zmieniają swój fenotyp w ten sposób, że stają się producentami
elementów macierzy pozakomórkowej, w tym głównie kolagenu typu I i II i elastyny oraz proteoglikanów (varsikan, biglikan i inne). Migrujące miocyty i produkowane przez nie elementy włókniste (kolagen, elastyna) wrastają w jądro lipidowe blaszki, a także otaczają je, tworząc
tak zwaną pokrywę blaszki miażdżycowej (ryc. 1.3.). Przyczynia się to do dalszego zwiększenia
objętości płytki (i ewentualnie wielkości stenozy) ale równocześnie elementy łącznotkankowe
utwardzają blaszkę i powodują, że jej pokrywa staje się odporna na pękanie;
6. Od pewnego etapu rozwoju blaszki, dochodzi do wrastania do niej drobnych naczyń mikrokrążenia. Odbywa się to prawdopodobnie pod wpływem czynników angiogenetycznych, których ekspresja w blaszkach jest duża (np. VEGF). Mogą być tego przynajmniej
3 konsekwencje: (i) rośnie powierzchnia, przez którą do blaszki mogę penetrować nowe
komórki zapalne, więc bogate unaczynienie może sprzyjać destabilizacji blaszki; (ii) może
ono promować systematyczne powiększanie się objętości blaszki, w podobnym mechanizmie jak to ma miejsce guzach nowotworowych (inhibitory angiogenezy zmniejszają rozmiary zmian miażdżycowych w eksperymentalnej miażdżycy) oraz (iii) mikronaczynia są
prawdopodobnie dość kruche, co skutkuje wynaczynieniami krwi do blaszki i powstawaniem wewnątrzblaszkowych zakrzepów. Istnieje propozycja, że te wynaczynienia są silnym
bodźcem do szybkiego powiększania się blaszki gdyż produkowany przez wynaczynione
płytki krwi czynnik PDGF (platelet derived growth factor) silnie promuje proliferacje komórek mięśni gładkich i produkcję przez nie elementów macierzy pozakomórkowej;
7. Od pewnego etapu rozwoju blaszki, dochodzi do odkładania się w niej złogów wapnia
i dalszego utwardzania się jej struktury. Komórkami odpowiedzialnymi za ten proces są
przekształcone komórki naczyniowych mięśni gładkich.
VII.3. Zmiany naczyniowe towarzyszące chorobie wieńcowej
W naczyniach dotkniętych miażdżycą występują przynajmniej trzy rodzaje zaburzeń naczyniowych, które w zaawansowanej postaci mogą być przyczyną niedokrwienia mięśnia sercowego i związanej z tym symptomatologii choroby niedokrwiennej serca. Chodzi o blaszki
miażdżycowe, dysfunkcję śródbłonkową oraz niezbyt dobrze poznane zaburzenia w naczyniach mikrokrążenia, które są przyczyną kardiologicznego zespołu X.
Blaszki miażdżycowe (tab. 7.1.) stają się źródłem objawów klinicznych, kiedy zwężają tętnicę (stenozy tętnic nasierdziowych są przyczyną stabilnej choroby wieńcowej) albo/i kiedy
z ich udziałem dochodzi do powstania lokalnego zakrzepu (aterotromboza) w jakimś obszarze naczyniowym.
160
Tabela 7.1. Blaszki miażdżycowe w tętnicach wieńcowych
• Mogą być obecne w angiograficznie prawidłowych tętnicach;
• Powstawanie stenozy wieńcowej jest wypadkową procesów: wzrostu blaszki i przebudowy
naczynia. Stenoza powstaje w wyniku niewydolności procesu przebudowy;
• Blaszki powodujące ekscentryczne stenozy są częstsze (~75%) niż te powodujące stenozy
koncentryczne (~25%);
• Wzrost blaszki (i stenozy) jest często procesem skokowym;
• Oceniane angiograficznie zaawansowanie stenozy wieńcowej nie pozwala przewidzieć ryzyka
wystąpienia OZW;
• Blaszki ulegają erozji i/lub pękają, kiedy są „niestabilne” a nie, kiedy powodują stenozę;
• O niestabilności blaszek decyduje ich szczególna budowa;
• W tym samym sercu mogą istnieć blaszki stabilne i niestabilne;
• Nie każda erozja/pęknięcie blaszki skutkuje wewnątrznaczyniowym zakrzepem;
• Nie każdy zakrzep jest źródłem OZW.
VII.3.1. Klasy stenoz i ich wpływ na symptomatologię stabilnej dusznicy
Objawy stabilnej dusznicy są najczęściej wtórne do obecności miażdżycowej stenozy
w nasierdziowej tętnicy wieńcowej. Nasilenie objawów zależy od stopnia zaawansowania stenozy (Klasy I –III) (omówienie w rozdz. III.5). O obrazie chorobowym decyduje także to czy
blaszka/stenoza jest koncentryczna czy ekscentryczna.
VII.3.2. Blaszki koncentryczne i ekscentryczne
O symptomatologii stabilnej choroby wieńcowej, obok wielkości stenozy (rozdz. VII.3.5)
decyduje to czy blaszka jest ekscentryczna czy koncentryczna. Blaszki koncentryczne są przyczyną ~25% stenoz wieńcowych – sztywnych i słabo reagujących na bodźce skurczowe i rozkurczowe. Są przyczyną wysiłkowej dusznicy charakteryzującej się stałą symptomatologią kliniczną
(niezmienna tolerancja wysiłku i stały próg niedokrwienia) i korzystną reakcja na β-blokery.
Blaszki ekscentryczne, obejmujące jedynie część obwodu naczynia, są odpowiedzialne za ~75%
stenoz wieńcowych. Część nieobjęta blaszką jest szczególnie wrażliwa na czynniki naczyniokurczące (np. ergonowina) gdyż czynność pokrywającego ją śródbłonka jest na ogół upośledzona.
W czasie wysiłku, zamiast rozkurczem, może ona reagować paradoksalnym skurczem, co, obok
stenozy, dodatkowo upośledza przepływ. Nasilenie tej naczynioskurczowej reakcji zmienia się
wraz ze zmianami czynności śródbłonka spowodowanymi czynnikami środowiska (dieta wysokotłuszczowa, palenie, brak ruchu etc.). Dlatego dusznica z udziałem blaszek ekscentrycznych
charakteryzuje się zmienną symptomatologią i zmienną tolerancji wysiłku.
VII.3.3. Ogniskowe vs. uogólnione rozprzestrzenienie zmian
Miażdżyca powstaje w wyniku działania czynników ryzyka, które swym wpływem obejmują całe łożysko naczyniowe. Towarzyszą temu przynajmniej dwie uogólnione zmiany naczyniowe, jakimi są: (i) dysfunkcja śródbłonkowa dotycząca całej tętniczej części układu naczyniowego, włączając w to naczynia mikrokrążenia (np. dysfunkcja śródbłonkowa w naczyniach mikrokrążenia) (rozdz. VII.3.5) oraz (ii) uogólnione pogrubienie warstwy wewnętrznej
tętnic, co jest widoczne jako wzrost stosunku grubości intima/media w dużych tętnicach
osobników z miażdżycą. Natomiast blaszki miażdżycowe, z niejasnych powodów, występują
jedynie w dyskretnych miejscach dużych i średnich tętnic.
161
Tradycyjna interpretacja tego faktu jest taka, że laminarny przepływ krwi i towarzysząca
temu siła ścinająca (rozdz. III) są czynnikami kształtującymi przeciw-zapalny, przeciw-krzepliwy i, tym samym, przeciw-miażdżycowy fenotyp śródbłonka. Okazuje się, że zaburzenia hydrodynamiczne w postaci zmniejszonej siły ścinającej i/lub przepływu burzliwego są szczególnie
duże w dużych tętnicach w miejscu odgałęzień, rozdwojeń i zagięć tych tętnic, czyli w miejscach
występowania blaszek miażdżycowych. Przyjmuje się, że efekty tych zaburzeń hydrodynamicznych sumując się z efektami czynników ryzyka, decydują o szczególnej lokalizacji blaszek [36].
Badania eksperymentalne sugerują, że obok dyskretnej lokalizacji i wielkości zmiany miażdżycowej, także stan stabilności blaszki zależy od rozkładu sił hydrodynamicznych
w naczyniach. W badaniach tych wykazano, że miejsca z wyraźnie zmniejszoną siłą ścinającą miały większe blaszki i ich budowa histologiczna była typowa dla blaszek niestabilnych.
Natomiast miejsca z przeważającym przepływem burzliwym (np. obszary za stenozą) miały
blaszki o fenotypie blaszek stabilnych (dużo kolagenu, mało składników lipidowych) [37].
Inna możliwość jest taka, że o specjalnej lokalizacji blaszek decydują jakieś lokalne różnice w biologii ściany naczyniowej. Dla przykładu, opisano różnice w embrionalnym mechanizmie rekrutacji komórek mięśni gładkich do różnych części drzewa naczyniowego. Na
poparcie tej koncepcji, wykazano, że przeszczepienie młodym zwierzętom segmentu aorty
brzusznej (odcinek szczególnie podatny na rozwój zmian miażdżycowych) do odcinka piersiowego (odporny na proces miażdżycowy) nie zapobiegało rozwojowi zmian. Dla kontrastu,
segment piersiowy przeszczepiony do brzucha nie rozwijał zmian [38]. Równie zagadkowa
pozostaje przyczyna częstego występowania u tych samych osobników (nawet w bezpośrednim sąsiedztwie) blaszek stabilnych i niestabilnych, z przebudową do- i odśrodkową.
VII.3.4. Przebudowa ściany naczyniowej
Wraz z powiększaniem się masy blaszki miażdżycowej, ściana tętnicy ulega przebudowie
dośrodkowej (negatywnej) lub odśrodkowej (pozytywnej). Dośrodkowa polega na tym, że zawartość blaszki wpukla się do światła tętnicy powodując jej zwężenie (stenozę) i ograniczenie
rezerwy wieńcowej w obszarze za stenozą, co może prowadzić do objawów stabilnej choroby
wieńcowej. Przebudowa odśrodkowa polega na tym, że zawartość blaszki „popycha” ścianę tętnicy na zewnątrz (rośnie jej zewnętrzna średnica, aż do powstawania zmian tętniakowatych),
co sprawia, że wewnętrzna średnica naczynia pozostaje niezmieniona. Ten typ przebudowy
jest korzystny z hydraulicznego punktu widzenia gdyż zapobiega powstawaniu stenozy. Warunkiem przebudowy odśrodkowej tętnicy jest jednak aktywacja procesu zapalnego w blaszce
i towarzysząca aktywacja metaloproteinaz. Oba te procesy są odpowiedzialne za destabilizację
blaszek miażdżycowych. Rzeczywiście, blaszki, którym towarzyszy przebudowa odśrodkowa
mają „niestabilną” budowę. U pacjentów ze stabilną chorobą wieńcową wykazano korelację
między stopniem odśrodkowej przebudowy (ocena przy użyciu wewnątrznaczyniowej ultrasonografii) i poziomem w surowicy markerów stanu zapalnego (rozpuszczalne cząstki adhezyjne
sVCAM-1, sICAM-1, sE-selektyna oraz hsCRP) [39]. Nie wiadomo, dlaczego u tej samej osoby
niektóre blaszki przechodzą przebudowę odśrodkową a inne dośrodkową. Sumując, nawet duża
blaszka miażdżycowa może być niewidoczna w badaniu angiograficznym. Negatywny wynik
tego badania nie pozwala wykluczyć obecności miażdżycy tętnic wieńcowych. Właśnie blaszki
niewidoczne angiograficznie są zwykle niestabilne i są częstą przyczyną OZW [40].
162
VII.3.5. Dysfunkcja śródbłonkowa
Konsekwencje dysfunkcji dla obszaru naczyniowego ze zmianami miażdżycowymi mogą
być trojakie:
1. W obecności dysfunkcji czynniki kurczące zdobywają przewagę nad czynnikami rozkurczającymi ścianę naczyniową, co może skutkować paradoksalnym skurczem ściany tętnicy związanym
z wysiłkiem, zwłaszcza w sąsiedztwie ekscentrycznych blaszek miażdżycowych (rozdz. VII.3.2).
Niemniej jednak, mechanizm nadwrażliwości naczyń na czynniki kurczące nie jest dokładnie poznany i może obejmować jakieś niezbyt dobrze poznane zmiany w mięśniówce gładkiej dużych
tętnic (wtórne lub niezależne od dysfunkcji śródbłonkowej). W okolicy blaszek ekscentrycznych
może dochodzić do kurczu naczynia i dławicy spoczynkowej. Określenie kurcz naczynia (spazm
naczyniowy) oznacza zwykle nagły lokalny skurcz dużej tętnicy wieńcowej, często całkowicie ją
zaciskający, niezwiązany z wysiłkiem fizycznym. Zjawisko to jest przyczyną dławicy Prinzmetala.
Wśród bodźców skurczowych wywołujących kurcz tętnicy w tej postaci dławicy wymienia się
noradrenalinę (uwalnianą z zakończeń nerwów współczulnych i działająca poprzez receptory
alfa) serotoninę (pochodzącą z aktywowanych płytek) oraz histaminę (uwalnianą z komórek
tucznych). Spazm związany jest z napływem jonów wapnia do komórek mięśni gładkich poprzez
kanały wapniowe, dlatego przeciwdziałają mu antagoniści kanału wapniowego.
2. Dysfunkcja jest czynnikiem ryzyka występowania OZW (rozdz. IX), co sugeruje, że może
skutkować destabilizacją blaszek miażdżycowych.
3. Zwiększone ryzyko występowania OZW u osób z dysfunkcją śródbłonkową może być także konsekwencją pro-zakrzepowego fenotypy „dysfunkcjonalnego” śródbłonka” , na który
składają się: (i) promocja aktywacji płytek; (ii) promocja aktywacji trombiny i wykrzepiania oraz (iii) zmniejszona aktywność fibrynolityczna śródbłonka (tab. 7.2.).
Tabela 7.2. Mechanizm przeciwkrzepliwego działania zdrowego śródbłonka i pro-krzepliwego działania śródbłonka z dysfunkcją
Fenotyp przeciw-krzepliwy
prawidłowego śródbłonka
1. Hamowanie płytek krwi
a. NO, prostacyklina;
b. nieadhezyjna powierzchnia;
2. Zapobieganie powstawaniu trombiny i,
tym samym, tworzeniu zakrzepu
a. siarczany heparanu składające się na
proteoglikany budujące glikokaliks komórek
śródbłonka;
b. trombomodulina związana z heparanem
i wiążąca trombinę;
c. inhibitor czynnika tkankowego związany
z heparanem;
Fenotyp pro-krzepliwy
śródbłonka z dysfunkcją
1. Osłabione hamowanie płytek krwi
a. rNO, rprostacyklina
b. promocja adhezji płytek -qekspresja
selektywny P i czynnika von Willebrandta;
c. utrata ciągłości śródbłonka;
2. Promocja produkcji trombiny i tworzenia
zakrzepu
a. destrukcja glikokaliksu;
b. qekspresja czynnika tkankowego;
3. Osłabione działanie fibrynolityczne
a. -qekspresja inhibitora t-PA (PAI-1);
b. rekspresja t-PA;
3. Promocja fibrynolizy
a. tkankowy i urokinazowy aktywator
plazminogenu (t-PA i u-PA);
163
Wiadomo obecnie, że dysfunkcja śródbłonkowa jest procesem ogólnoustrojowym i że
obok dużych naczyń dotyka także naczyń mikrokrążenia. Jest coraz więcej faktów świadczących o tym, że takim czynnikom ryzyka jak nadciśnienie, hypercholesterolemia, cukrzyca
i otyłość, obok dysfunkcji śródbłonkowej (rozdz. IX), towarzyszą uogólnione zmiany fenotypu śródbłonka, których konsekwencją są: (i) zwiększona adhezja leukocytów do śródbłonka; (ii) „rozszczelnienie” bariery śródbłonkowej; (iii) zwiększona rekrutacja płytek krwi do
śródbłonka i zwiększona aktywność procesu krzepnięcia i inne [35]. Możliwą konsekwencją tych uogólnionych zmian śródbłonkowych jest to, że w ich obecności narządy, w których
wystąpiło niedokrwienie gorzej to niedokrwienie tolerują [35]. Dla przykładu u pacjentów
z zawałem leczonych przy pomocy plastyki wieńcowej, częstość występowania no-reflow phenomenon w okresie reperfuzji była większa u osób z wyjściowo wyższymi poziomami hsCRP
w surowicy [41].
VII.4. Ostre zespoły wieńcowe (OZW)
VII.4.1. Badania autopsyjne u osób zmarłych w wyniku OZW
Nagła śmierć sercowa; Badania autopsyjne sugerują, że prawdopodobną przyczyną ~50%
przypadków nagłej śmierci sercowej są nadżerki śródbłonka na powierzchni blaszki (plaque
erosion, plaque fissure). U pozostałej części badanych stwierdzano albo pęknięcia blaszek albo
brak jakichkolwiek uszkodzeń blaszek (sugestia – skurcz naczyniowy jako możliwa przyczyna arytmii i zgonu) [42]. Nadżerki prowokowały zwykle powstawanie kruchego płytkowego
zakrzepu, luźno związanego z podłożem. Stwarza to prawdopodobnie warunki do odrywania
się z jego powierzchni agregatów płytkowych, które zatykając naczynia mikrokrążenia wieńcowego, stają się przyczyną mikrozawałów, a tym samym niestabilności elektrycznej serca
i groźnych zaburzeń rytmu, prowadzących do zgonu.
Niestabilna dusznica; Za tę postać OZW odpowiedzialne są zwykle uszkodzenia ekscentrycznych blaszek miażdżycowych, a skrzeplina z nimi związana zwykle penetruje do światła
tętnicy wieńcowej i w różnym stopniu ogranicza przepływ wieńcowy, ale go całkowicie nie
blokuje. Na „niestabilność” zespołu składają się przynajmniej trzy elementy:
(1) Powstawanie skrzepliny jest wypadkową równocześnie zachodzących procesów wykrzepiania i fibrynolizy. Dlatego wielkość wewnątrznaczyniowej skrzepliny, nawet w krótkich
odstępach czasu, na przemian rośnie i maleje, co skutkuje niedokrwieniem o zmieniającym się w czasie nasileniu [43];
(2) Ponieważ za niestabilną anginę odpowiedzialne są głównie ekscentryczne blaszki, fragment
ściany naczynia nie objęty zmianą miażdżycową może się z różnym nasileniem kurczyć,
między innymi pod wpływem uwalnianych z aktywowanych płytek substancji o działaniu
naczyniokurczącym (tromboksan, seroronina) i dodatkowo ograniczać drożność tętnicy;
(3) Powierzchnia zakrzepu penetrującego do światła tętnicy jest pokryta warstwą zaktywowanych płytek (miarą tej aktywacji jest wzrost ekspresji na ich powierzchni receptora GP IIb/
IIIa i wobec tego ich podatność na działanie inhibitorów tego receptora). Agregaty tych płytek mogą się odrywać od powierzchni zakrzepu i w ten sposób stanowią materiał zatorowy
dla naczyń mikrokrążenia. Obecność troponiny T w surowicy pacjentów z niestabilną dusznicą jest użytecznym wskaźnikiem powstawania mikrozawałów u tej grupy chorych.
164
Zawały z uniesieniem odcinka ST (STEMI); Przyczyną są na ogół zakrzepy, które całkowicie i nagle zamknęły światło tętnicy wieńcowej. W efekcie martwica zawałowa rozwija
się stosunkowo szybko (poniżej 12 godzin). W 75% przypadków zawał taki jest wynikiem
pęknięcia blaszki miażdżycowej, a w 25% nadżerki śródbłonka [42]. Badania angiograficzne
wykazują, że zakrzep prowokujący zawał jest dynamiczną strukturą, i że w kilkanaście godzin
po uszkadzającym zamknięciu światła tętnicy, w około 50% przypadków dochodzi do lizy
zakrzepu i spontanicznego udrażniania się tętnicy zawałowej [44].
Zawały bez uniesienia odcinka ST (NSTEMI); Mechanizm powstawania jest prawdopodobnie bardziej skomplikowany, a rozwój martwicy bardziej rozciągnięty w czasie niż
w zawałach STEMI. Zawały NSTEMI są często konsekwencją niestabilnej dławicy. Ważnym
czynnikiem decydującym o ich powstawaniu jest również to, że obszary nimi objęte (w przeciwieństwie do obszarów ulegających zawałowi STEMI) mają zwykle dość dobrze rozwinięte
krążenie oboczne. Zawał tzw. niepełnościeny jest zwykle wynikiem fuzji kilku mniejszych
obszarów martwicy powstałych wskutek okresowego niedokrwienia i reperfuzji. Martwica
jest również wynikiem fuzji mikrozawałów będących skutkiem zatorów mikrokrążenia spowodowanych przez agregaty płytkowe. O ile zawały STEMI rozwijają się stosunkowo szybko,
śmierć poszczególnych fragmentów niepełnościennego zawału może dzielić nawet okres 7–10
dni [42]. Przytoczona powyżej charakterystyka dotyczy jednak tylko tych sytuacji, w których
zawał miał przebieg „naturalny”, to znaczy nie był leczony przy pomocy reperfuzji lub reperfuzja okazała się nieskuteczna. W dobie leczenia reperfuzyjnego, źródłem zawałów NSTEMI
mogą być również niedokrwienia, które w warunkach „naturalnych” skończyły by się zawałem STEMI, ale zanim do tego doszło, zostały poddane skutecznej reperfuzji.
VII.4.2. Czteroczynnikowy model mechanizmu OZW
Wyniki badań autopsyjnych sugerują, że prawdopodobna kaskada procesów prowadzących do powstania OZW wygląda jak pokazuje ryc. 7.6. [42;45-49].
Ryc. 7.6. Historia naturalna ostrych zespołów wieńcowych, tj. niestabilnej anginy, nagłej śmierci sercowej, zawału NSTEMI oraz zawału STEMI.
165
Konkluzja z tych danych oraz obserwacji klinicznych jest taka, że (i) uszkodzenie blaszki
nie jest warunkiem wystarczającym do wystąpienia OZW (10–15% uszkodzeń bez OZW)
oraz (ii) takie samo uszkodzenie blaszki i zakrzep skutkują różnie nasilonym występowaniem
OZW (np. nagłe śmierci sercowe występują tylko u części pacjentów).
Te i inne obserwacje dowodzą, że mechanizm OZW można traktować w kategoriach „katastrofy” naczyniowej i że jak zawsze w przypadku katastrof, warunkiem wystąpienia OZW
jest splot kilku nieszczęśliwych elementów. Aktualna propozycja zakłada, że chodzi tutaj
o współgrę przynajmniej czterech elementów, dwóch lokalnych związanych z właściwościami
samej blaszki oraz dwóch zewnętrznych w stosunku do blaszki i w znacznej mierze ogólnoustrojowych [50;51] (ryc. 7.7.). Czynniki, o których mowa to:
1. Szczególna podatność blaszki miażdżycowej na uszkodzenie; Blaszki różnią się skłonnością
do pękania i prowokowania wewnątrznaczyniowego zakrzepu. Źródłem OZW są raczej
tzw. niestabilne blaszki (vulnerable plaque, unstable plaque, high-risk plaque, dangerous
plaque, thrombosis-prone-plaque) [50], wyróżniające się szczególną anatomiczną strukturą
i składem histologicznym, a nie faktem, że powodują duże czy małe zwężenia [42,45-49].
Ryc. 7.7. Czteroelementowy mechanizm OZW i prawdopodobne mechanizmy protekcyjnego działania
aspiryny, β-blokerów, inhibitorów konwertazy (ACE-I) i statyn, czyli leków z klinicznie udokumentowaną zdolnością do zmniejszania ryzyka występowania OZW. Zwraca uwagę plejotropowe działanie
tych leków.
2. Lokalna „skłonność” blaszki i jej najbliższego otoczenia do indukowania zakrzepu („niestabilny układ krzepnięcia”, vulnerable blood); Powstawanie zakrzepu i jego ostateczna
wielkość są wypadkową czterech współistniejących czynników: (a) rozległości uszkodzenia
blaszki miażdżycowej; (b) aktywności procesów adhezji i agregacji płytek; (c) aktywności procesu krzepnięcia (koagulacji) przez obecne w blaszce aktywatory krzepnięcia (np.
czynnik tkankowy) oraz (d) aktywności procesu fibrynolizy, zależnej między innymi od
166
śródbłonkowej produkcji tkankowego aktywatora plazminogenu (tPA) i inhibitora tego
aktywatora (PAI-1). Uszkodzenie blaszki miażdżycowej jest prawdopodobnie warunkiem
koniecznym, ale niewystarczającym do powstania zakrzepu zatykającego tętnicę wieńcową
(ryc. 7.6.) [46-49].
3. Obecność czynnika/bodźca uszkadzającego blaszkę w takim stopniu, by możliwy stał się
kontakt krwi z trombogennym wnętrzem blaszki. W tym kontekście podnoszona jest rola
czynników hemodynamicznych (siła ścinająca, ciśnienie krwi etc.), ale możliwy jest także
scenariusz, w którym destrukcja pokrywy blaszki jest wynikiem jej trawienia przez wewnątrzblaszkowe metaloproteinazy (rozdz. VII.4.3).
4. Zespołu ogólnoustrojowych czynników określanych jako „niestabilny pacjent” (vulnerable patient). Chodzi tutaj o przynajmniej dwie obserwacje: (i) podobne katastrofy naczyniowe prowadzą do nagłej śmierci sercowej tylko u niektórych osób, co każe przypuszczać,
że ich serca mają większą łatwość reagowania arytmią na zaburzenia związane z katastrofą
naczyniową (niestabilne miokardium, vulnerable myocardium); (ii) czynnikiem ryzyka
występowania OZW są zwiększone poziomy markerów zapalenia we krwi (np. CRP), co
sugeruje, że ogólnoustrojowy odczyn zapalny jest czynnikiem destabilizującym blaszki
miażdżycowe.
Z takiego wieloelementowego modelu wynikają dwie ważne konsekwencje: (i) ryzyko
występowania OZW powinno maleć zarówno w rezultacie działań stabilizujących blaszki, jak
i wymierzonych w którykolwiek z pozostałych elementów modelu; (ii) fakt, że jakieś działanie
terapeutyczne zmniejsza ryzyko występowania OZW nie oznacza, że to działanie ma związek
ze stabilizacją blaszek miażdżycowych.
Dyskutowany model mechanizmu OZW jest w znacznej mierze konstrukcją hipotetyczną wymagającą w wielu punktach ostatecznej weryfikacji, a z tym są nieprzezwyciężalne
obecnie trudności. Udokumentowane działanie zmniejszające ryzyko występowania OZW
mają: aspiryna, beta-blokery, inhibitory konwertazy i statyny. Wszystkie te interwencje mają
jednak liczne działania plejotropowe (ryc. 7.7.) i można spekulować, że właśnie ta wielokierunkowość działania decyduje o ich wyjątkowej skuteczności. Z efektów klinicznych tych
interwencji nie wynikają natomiast wnioski ani co do ich mechanizmu działania, ani co do
mechanizmu OZW w ogólności. Postęp w tej ostatniej kwestii przyniosą prawdopodobnie
dopiero badania z interwencjami o wybiórczym działaniu przeciw-płytkowym, przeciw-krzepliwym, przeciw zapalnym etc., jeżeli takie zostaną kiedyś wynalezione.
VII.4.2.1. Niestabilna blaszka miażdżycowa
Postęp, jaki się ostatnio dokonał w klinice choroby wieńcowej zawdzięczamy głównie rozwojowi kardiologii interwencyjnej. Z natury rzeczy, uwaga kardiologa interwencyjnego jest zogniskowana na zmianach miażdżycowych, które powodują znaczną stenozę tętnic wieńcowych.
Pochodną tego jest przekonanie, że nasilenie objawów choroby wieńcowej rośnie wraz z zaawansowaniem procesu miażdżycowego mierzonego rozmiarami stenozy wieńcowej. Reguła ta
rzeczywiście obowiązuje w przypadku symptomatologii stabilnej choroby wieńcowej. Nie jest
pewne czy dotyczy OZW [52]. Badania, w których analizowano wyniki badań arteriograficznych uzyskanych u tych samych osób przed i po OZW sugerują, że przyczyną większości OZW
jest pękanie małych „niestenotycznych” blaszek miażdzycowych. Dla kontrastu, badania au167
topsyjne (z konieczności dotyczące osób, które zmarły) oraz badania z użyciem ultrasonografii
wewnątrznaczyniowej i tomografii tętnic wieńcowych sugerują, że raczej duże blaszki (z rozsianymi ogniskami wapnienia), a nie małe, są najczęstszymi „winowajcami” OZW.
Trzy rodzaje obserwacji klinicznych i anatomopatologicznych dowodzi, że ryzyko wystąpienia OZW jest pochodną „niestabilności” blaszki a nie faktu, że powoduje ona taką czy
inną stenozę:
1. Badania autopsyjne wykazują, że w tętnicach wieńcowych osób, które zmarły z powodu
OZW znajduje się zwykle wiele blaszek miażdżycowych o podobnych rozmiarach. Jednakże za wystąpienie OZW odpowiedzialne jest zazwyczaj uszkodzenie tylko jednej z nich,
o charakterystycznej strukturze wewnętrznej [53]. Sugeruje to, że blaszka ta musiała być
szczególnie podatna na uszkodzenie, czyli niestabilna;
2. Badania prospektywne u osób, którym przed i po OZW wykonywano seryjne badania angiograficzne wykazały, że chociaż blaszki powodujące znaczne zwężenia naczyń były procentowo bardziej narażone na pęknięcie, to jednakże zmiany niepowodujące zwężeń, lub
małe zwężenia (<50% stenozy), częściej prowadziły do OZW, ze względu na ich duży udział
procentowy w całkowitej liczbie blaszek w całym sercu [54]. Podobne wnioski płyną z prospektywnych badań osób z chorobą wieńcową, w których morfologię blaszek oceniano przy
pomocy wewnątrznaczyniowej ultrasonografii [55]. Blaszki bez stenozy częściej prowadzą
do OZW prawdopodobnie, dlatego że mają proporcjonalnie większą zawartości składników
lipidowych, które je destabilizują [56]. Inny powód to ten, że krążenie oboczne jest zwykle
lepiej rozwinięte w sąsiedztwie dużych a nie małych stenoz wieńcowych [57] i wobec tego
skrzeplina zamykająca tętnicę z dużą stenozą wywołuje mniejsze niedokrwienie;
3. Różne interwencje lecznicze, o których wiadomo, że zmniejszają występowanie OZW (np.
statyny, ACE-inhibitory), nie zmniejszają wielkości miażdżycowych zwężeń tętnic wieńcowych [58]. Sugeruje to, że interwencje te zmniejszają ryzyko występowania OZW poprzez
„stabilizację” istniejących blaszek a nie wpływ na proces ich wzrostu.
Tabela 7.3. Charakterystyka blaszek miażdżycowych odpowiedzialnych za powstawanie
ostrych zespołów wieńcowych
Struktura
Najczęściej ekscentryczne
Duże jądro lipidowe (> 40–50% objętości blaszki)
Cienka pokrywa łącznotkankowa
Zmniejszona zawartość kolagenu
Histologia
Cechy komórkowe przewlekłego zapalenia
Zwiększona gęstość i aktywność makrofagów w blaszce
Akumulacja limfocytów T w pobliżu miejsca pęknięcia blaszki
Zwiększona liczna i aktywność komórek tucznych
Zmniejszona gęstość komórek mięśni gładkich
Zwiększona neowaskularyzacja
Biochemia
Zwiększona ekspresja metaloproteinaz
Zwiększona ekspersja czynnika tkankowego
Obecność markerów biochemicznych aktywacji procesu zapalnego np. cytokiny, interleukiny
168
Razem wziąwszy dane te sugerują, że proces wzrostu masy blaszki i stan jej „niestabilności” można traktować jako dwa odrębne zjawiska patofizjologiczne wymagające innego traktowania terapeutycznego.
Z badań, głównie, autopsyjnych wynika, że typowe cechy blaszek „winowajców” OZW
to: (i) cienka pokrywa łącznotkankowa; (ii) duże miękkie jądro lipidowe oraz (iii) duża zawartość komórek zapalnych [42]. Żadna z tych cech nie koreluje ani z rozległością blaszki, ani
z wielkością wywoływanej przez nią stenozy [58]. Te i inne cechy blaszek „winowajców” (tab.
7.3.) potwierdzają hipotezę, że blaszki stają się niestabilne wtedy, kiedy toczący się w nich
proces zapalny zyskuje przewagę nad mechanizmami reparacyjnymi (ryc. 7.8.).
Aktywny proces zapalny w blaszkach destabilizuje je gdyż sprzyja: (i) powiększaniu się
jądra lipidowego (bo duża jest wtedy liczba umierających komórek piankowatych) oraz (ii)
trawieniu pokrywy łącznotkankowej przez metaloproteinazy produkowane przez makrofagi
(ryc. 7.8.). Jak wykazują badania morfologiczne, właśnie: duże jądro lipidowe (>50% objętości blaszki), duża zawartość w blaszce makrofagów, czynnika tkankowego i metalloproteinaz
(produkowanych przez makrofagi) oraz cienka pokrywa łącznotkankowa to typowe elementy
budowy blaszek odpowiedzialnych za powstawanie OZW [42].
Ryc. 7.8. Stabilność blaszki miażdżycowej jako wypadkowa efektów procesu zapalnego i mechanizmów
reparacyjnych prowadzących do separacji i utwardzenia struktury blaszki
Wymiary i konsystencja jądra lipidowego różnią się pomiędzy różnymi blaszkami u tego
samego osobnika. Blaszka jest szczególnie narażona na pęknięcie, gdy jej jądro lipidowe zajmuje ponad 40% jej objętości. Jądro składa się przede wszystkim z cholesterolu (wolnego i zestryfikowanego) pochodzącego z LDL, z domieszką żywych komórek piankowatych. W temperaturze ciała, jądro lipidowe ma konsystencję pasty do zębów, z tym, że estry cholesterolu
mają konsystencję bardziej płynną niż krystaliczny cholesterol. Obniżanie stężenia cholesterolu we krwi zmniejsza ryzyko występowania ostrych epizodów wieńcowych, prawdopodobnie poprzez zmniejszanie napływu LDL do blaszki i zmniejszanie takich niekorzystnych tego
konsekwencji jak: dysfunkcja śródbłonka, aktywacja makrofagów i powiększanie się jądra
lipidowego [59].
169
Jądro lipidowe blaszki miażdżycowej jest ponadto ważnym magazynem czynnika tkankowego, który produkowany głównie przez makrofagi, jest silnym aktywatorem procesu
krzepnięcia.
Pokrywa łącznotkankowa składa się przede wszystkim z kolagenu, elastyny i pojedynczych komórek mięśni gładkich. Kolagen wytwarzany jest przez stymulowane cytokinami
i czynnikami wzrostowymi komórki mięśni gładkich ściany naczynia. W blaszkach ekscentrycznych, pokrywa jest najcieńsza w obszarze ich przylegania do niezmienionej części naczynia. Badania modelowe i bezpośrednie pomiary dowodzą, że właśnie w tych miejscach działają na blaszkę największe siły rozrywające jej pokrywę. W tym miejscu na ogół zlokalizowane
są również skupiska makrofagów (ryc. 7.9.).
Blaszki odpowiedzialne za OZW zawierają zazwyczaj więcej makrofagów niż te odpowiedzialne za stabilną dusznicę bolesną. Komórki zapalne naciekają blaszkę przede wszystkim w miejscu jej połączenia z wolną ścianą naczynia, czyli w okolicy najbardziej narażonej
na mechaniczne rozerwanie przez siły związane z czynnikami hemodynamicznymi. Okolica
ta jest także szczególnie narażona na uszkadzające działanie czynników pochodzących z jej
wnętrza od komórek zapalnych. Udział komórek zapalnych w osłabianiu struktury pokrywy
blaszek może być trojaki. Makrofagi, poprzez produkcję metaloproteinaz (produkowane są
nieaktywne pro-enzymy), przyczyniają się do degradacji kolagenu i elastyny. Limfocyty T,
poprzez produkcję interferonu γ, hamują produkcję kolagenu przez komórki mięśni gładkich.
Interferon γ jest dodatkowo silnym aktywatorem makrofagów [13]. Natomiast aktywowane
komórki tuczne uwalniają takie enzymy proteolityczne, jak tryptaza i chymaza, które poprzez
działanie na pro-metaloprotinazy biorą udział w aktywacji tych enzymów. Nasilona aktywność komórek zapalnych w blaszce, indukuje również apoptozę, co z jednej strony prowadzi do uwalniania lipidów z obumarłych makrofagów i wzrostu objętości jądra lipidowego,
a z drugiej strony do zmniejszenia liczby komórek mięśni gładkich i osłabienia pokrywy łącznotkankowej (ryc. 7.9.).
Ryc. 7.9. Czynniki hemodynamiczne i związane z procesem zapalnym w blaszce miażdżycowej powodujące uszkodzenie pokrywy blaszki i stwarzające warunki do powstawania zakrzepu naczyniowego.
170
VII.4.2.2. Niestabilny układ krzepnięcia
Badania kliniczne sugerują, że czynnikami ryzyka występowania OZW są poziomy we
krwi różnych biochemicznych markerów zwiększonej krzepliwości krwi, w tym zwiększone
poziomy czynnika VII, fibrynogenu, czynnika von Willebranda, lipoproteiny A, homocysteiny i PAI-1 oraz obniżone poziomy t-PA.
Przynajmniej trzy elementy sprzyjają powstawaniu zakrzepu w okolicy uszkodzonej
blaszki miażdżycowej:
1. Miażdżycy zwykle towarzyszy uogólniona dysfunkcja śródbłonkowa, ale jest ona szczególnie nasilona w sąsiedztwie zmian miażdżycowych. Konsekwencją dysfunkcji jest tzw.
pro-zakrzepowy fenotyp śródbłonka (tab. 7.2.);
2. Uogólnionemu odczynowi zapalnemu, jaki może towarzyszyć CNS, a także otyłości, cukrzycy typu II i innym czynnikom ryzyka towarzyszy wzrost poziomu we krwi białek ostrej
fazy zapalenia, w tym fibrynogenu (substrat trombiny);
3. Uszkodzenie blaszki miażdżycowej jest bodźcem inicjującym proces powstawania zakrzepu za pośrednictwem dwóch współpracujących z sobą procesów: (i) adhezji i agregacji płytek oraz (ii) krzepnięcia krwi (koagulacji) (ryc. 7.10.).
W wyniku przerwania ciągłości śródbłonka zostają odsłonięte różne substancje (np.
kolagen, czynnik von Willebranda), które są ligandami dla receptorów płytek krwi. Płytki
przyczepiają się do tych substancji jedną warstwą (adhezja) i w wyniku tego procesu ulegają
aktywacji. Aktywacja obejmuje:
1. ekspresję na powierzchni płytek dużej liczby zaktywowanych integryn typu IIb/IIIa (GP
IIb/IIIa), które mają zdolność do wiązania fibrynogenu, i których obecność umożliwia
agregacje płytek, czyli powstawanie agregatów złożonych z płytek i fibrynogenu;
2. uwalnianie z ziarnistości płytkowych licznych substancji (tromboksan A2, ADP, czynnik
von Willebranda, czynnik V krzepnięcia, płytkopochodny czynnik wzrostu), które aktywują inne płytki krwi (dzięki czemu proces ich aktywacji może się szerzyć) a także kaskadę
krzepnięcia krwi.
Szczególnie korzystne warunki dla adhezji i agregacji płytek stwarzają nadżerki śródbłonka. Krzepnięcie jest w takich wypadkach słabiej aktywowane. Dlatego nadżerki skutkują
powstawaniem luźno związanej z podłożem, kruchej skrzepliny, która stwarza szczególnie
korzystne warunki dla odrywania się z jej powierzchni agregatów płytkowych.
Różnie nasilone pękanie pokrywy blaszki ma dwie konsekwencje: (i) miażdżycowa zawartość jej wnętrza i elementy pokrywy mogą przedostawać się do światła naczynia, gdzie stają
się elementem materiału zatorowego oraz (ii) masy miażdżycowe, bogate w czynnik tkankowy
aktywujący trombinę, stwarzają warunki dla adhezji i agregacji płytek i aktywacji krzepnięcia
[60,61]. Powstający zakrzep jest zwykle jednym końcem głęboko zakotwiczony we wnętrzu
pękniętej blaszki, a drugim penetruje do światła naczynia. Jego powstawanie ma trzy etapy:
1. w wyniku kontaktu krwi z wnętrzem lipidowym blaszki, w jej wnętrzu tworzy się zakrzep
zawierający głównie płytki i częściowo erytrocyty;
2. w wyniku działania czynnika tkankowego dochodzi do aktywacji czynnika VII krzepnięcia,
powstania aktywnej postaci aktywatora protrombiny, aktywacji trombiny i powstawania
włókien fibryny. W efekcie na skrzepie płytkowym odkłada się zbity zakrzep fibrynowy,
wypełniający wolne miejsca w uszkodzonej blaszce i otaczający okolicę jej pęknięcia;
171
Ryc. 7.10. Wielkość zakrzepu fibrynowego jest wypadkową powstawania fibryny z fibrynogenu pod
wpływem trombiny (koagulacja) oraz degradacji fibryny przez plazminę (fibrynoliza). Trombina i plazmina powstają jako nieaktywne pro-enzymy podlegające procesom kontrolowanej aktywacji. Czynnikom ryzyka OZW towarzyszą: (i) stan zwiększonej gotowości płytek do aktywacji (nie pokazane) i (ii)
krwi do krzepnięcia (zwiększona ekspresja substratu, fibrynogenu, i aktywatorów trombiny, np. czynnika tkankowego, TF) oraz (iii) zmniejszona aktywność fibrynolityczna (zmniejszony stosunek t-PA/
PAI-1). Uszkodzenie blaszki promuje adhezję i aktywację płytek oraz aktywację trombiny i krzepnięcia
via TF i mediatory płytkowe.
3. następuje odkładanie się luźniejszego fibrynowego zakrzepu, który w różnym stopniu wypełnia światło naczynia i ogranicza jego drożność. Dopiero ta trzecia warstwa zakrzepu jest
głównym obiektem działania środków fibrynolitycznych stosowanych w leczeniu zawału.
Konsekwencje kliniczne obecności wewnątrz-wieńcowego zakrzepu zależą od jego wielkości
i dynamiki narastania. W najkorzystniejszym przypadku, gdy przeważają procesy o charakterze fibrynolitycznym i naprawczym, zakrzep nawet, jeżeli jest głęboko zakotwiczony wewnątrz
blaszki, tylko w niewielkim stopniu penetruje do światła naczynia i nie ogranicza przepływu.
Taki zakrzep zostaje szybko pokryty warstwą komórek śródbłonka i staje się elementem struktury blaszki miażdżycowej. Blaszka szybko się powiększa, co stanowi prawdopodobną przyczynę
obserwowanego w klinice „skokowego” nasilenia się dynamiki rozwoju stabilnej choroby wieńcowej i/lub, stwierdzanego angiograficznie, nagłego wzrostu rozmiaru stenozy wieńcowej. Scenariusz ten potwierdza obecność elementów skrzepliny w strukturze blaszek miażdżycowych [62].
VII.4.2.3. Bodziec uszkadzający
Przepływowi krwi w tętnicach towarzyszą siły, które powodują: (1) rozrywanie ściany
naczynia (tzw. naprężenia okrężne powodujące pęknięcie ściany równoległe do długiej osi
naczynia i naprężenia podłużne powodujące pęknięcie poprzeczne) oraz (2) odwarstwianie
172
śródbłonka i ewentualnie głębszych warstw ściany naczynia (siła ścinająca związana z tarciem
lepkiej krwi o powierzchnię naczynia) [63]. Bezpośrednie pomiary i symulacje komputerowe sugerują, że w obecności ekscentrycznej blaszki z jądrem lipidowym bardzo wzrasta naprężenie okrężne w miejscu, gdzie pokrywa blaszki łączy się z niezmienioną częścią tętnicy,
a więc tam gdzie pokrywa blaszki jest najcieńsza [64]. Rzeczywiście, właśnie w tych miejscach najczęściej pękają blaszki odpowiedzialne za występowanie OZW. Nagły wzrost, którejś z wymienionych sił związany ze wzrostem ciśnienia krwi, rzutu minutowego czy rytmu
serca może stać się bezpośrednim bodźcem uszkadzającym blaszkę. Podobnie, powtarzające
się rozciągania, zginania czy skręcania tętnic związane z ruchami serca mogą być przyczyną
„zmęczenia materiału”, związanego z tym osłabienia struktury pokrywy blaszki i jej spontanicznego pękania [45] (ryc. 7.9.).
Częstość występowania OZW wykazuje charakterystyczny rozkład dobowy, ze wzrostem
częstości w godzinach porannej aktywności. W godzinach porannych występują także wzrosty
ciśnienia krwi, częstości akcji serca, aktywności układu współczulnego, skłonności do agregacji
płytek i krzepnięcia krwi [65]. Wiadomo także, że występowanie OZW ma często związek z poprzedzającym wysiłkiem fizycznym, aktywnością seksualną, czy różnymi formami pobudzenia
psychicznego. Stąd przekonanie, że czynnikiem uszkadzającym blaszkę może być jakiś, zewnętrzny w stosunku do blaszki, czynnik hemodynamiczny związany z aktywacją układu współczulnego (np. wzrost ciśnienia krwi, wzrost siły ścinającej wtórny do przyspieszenia przepływu krwi).
Z drugiej strony jednak, uszkodzenie blaszki może być także wynikiem działania czynników pochodzących z jej wnętrza. W tym kontekście najczęściej podnoszona jest rola różnych
mediatorów i efektorów procesu zapalnego toczącego się w niestabilnej blaszce (np. aktywacja
metaloproteinaz).
Fakt, że β-blokery zmniejszają występowanie OZW może mieć związek z ich działaniem
hemodynamicznym (ryc. 7.7.). Inne możliwe mechanizmy to bezpośrednie działanie na mechanizm arytmogenezy, działanie przeciw-płytkowe, działanie przeciw-krzepliwe (zmniejszają poziom fibrynogenu) i pro-koagulacyjne (zwiększają śródbłonkowa produkcję t-PA
i zmniejszają produkcję PAI-1 (ryc. 7.10.) i być może bezpośrednie działanie przeciw-miażdżycowe (obecne w zwierzęcych modelach miażdżycy).
VII.4.2.4. Systemowe zapalenie i „niestabilny” pacjent
Fakt, że podstawowym składnikiem blaszek miażdżycowych są komórki piankowate, czyli komórki wrodzonej odporności makrofagi obładowane cholesterolem, wyraźnie dowodzi
równoczesnego udziału procesu zapalnego i zaburzeń lipidowych w mechanizmie miażdżycy.
Znajduje to odzwierciedlenie w danych klinicznych pokazujących, że zarówno markery lipidowe (pozom cholesterolu) jak i zapalne (zwłaszcza poziom CRP) informują o ryzyku sercowo-naczyniowym pacjentów i wobec tego mogą służyć jako „przewodnik” leczenia.
Rolę zaburzeń lipidowych można podsumować w następujących punktach:
1. Zaawansowanie zmian miażdżycowych obserwowanych u płodów ściśle koreluje z poziomem cholesterolu u matek w czasie ciąży, co wskazuje na bardzo pierwotną rolę zaburzeń
lipidowych w patogenezie miażdżycy (rozdz. I.6);
2. Nie ma innego sposobu uzyskania eksperymentalnego modelu miażdżycy, jak poprzez modyfikowanie profilu lipoproteinowego zwierzęcia eksperymentalnego [66];
173
3. Badania epidemiologiczne wskazują na ścisły związek między poziomem LDL i/lub cholesterolu we krwi i występowaniem choroby wieńcowej [67];
4. Leczenie hipolipemizujące zmniejsza ryzyko zdarzeń sercowo-naczyniowych. Dobitnie pokazuje to niedawna metaanaliza 26 dużych randomizowanych badań klinicznych obejmujących w sumie 170 tys. pacjentów [68]. Okazało się, że redukcja frakcji LDL cholesterolu przy
pomocy dużych dawek statyn, także poniżej poziomu uznawanego dotychczas za optymalny
(1,8 mmol/l lub 70 mg/dl), bezpiecznie i w sposób ciągły zmniejszała ryzyko OZW, udaru
mózgu i zabiegu rewaskularyzacji. Nie zaobserwowano żadnej dolnej granicy poziomu LDL,
przy której pacjenci nie odnosiliby korzyści zdrowotnych. Dla całej analizowanej populacji
ustalono zależność, że każde obniżenie poziomu cholesterolu LDL o 1 mmol/l, niezależnie
od jego wyjściowego poziomu, redukuje ryzyko zdarzeń sercowo-naczyniowych o ~20%, co
sugeruj, że redukcja cholesterolu o 2–3 mmol/l może zmniejszyć to ryzyko o 40–50%.
Udział systemowego zapalenie (ocenianego w oparciu o pomiar markerów zapalenia we
krwi) w mechanizmie wydarzeń sercowo naczyniowych ilustrują następujące fakty:
1. Wykazano wśród osobników pozornie zdrowych i w różnych grupach pacjentów, że zwiększony poziom we krwi CRP jest czynnikiem ryzyka wydarzeń sercowo-naczyniowych
(obszerna literatura w ref. [12]), co sugeruje, że systemowy odczyn zapalny lub CRP, per
se, powodują destabilizację blaszek miażdżycowych skutkującą występowaniem wydarzeń
sercowo-naczyniowych;
2. Leczenie zmniejszające poziom CRP zmniejsza ryzyko sercowo-naczyniowe. Wskazuje
na to niedawne wieloośrodkowe badanie JUPITER, do którego włączono osoby bez wcześniejszej historii choroby wieńcowej (n ≈17 800), bez hiperlipidemii (cholesterol LDL <
3,4 mmol/L lub <130 mg/dl), ale z podwyższonym poziomem CRP (CRP >20 nmol/L lub
>2 mg/l). Połowa pacjentów otrzymywała rosuwastatynę (20 mg/dobę) a połowa placebo.
[69,70]. Badanie wykazało, że statyna zmniejszyła ryzyko sercowo-naczyniowe oraz śmiertelność nawet u osób z cholesterolem LDL znacznie poniżej poziomu aktualnie uważanego
za wskazanie do leczenia. Interpretacja tego badania jest taka, że statyny redukują ryzyko
sercowo-naczyniowe zarówno poprzez redukcję poziomu cholesterolu LDL jak i poprzez
efekt przeciwzapalny (prawdopodobnie niezależny od LDL).
3. Wykazano dodatnią korelację między poziomem CRP i innych markerów zapalenia oraz
stopniem odśrodkowej przebudowy miażdżycowo zmienionych tętnic wieńcowych (ocena
przy pomocy wewnątrznaczyniowej ultrasonografii). Skądinąd wiadomo, że przyczyną odśrodkowej przebudowy jest aktywny proces zapalny w blaszce (rozdz. VII.3.4).
Niejasny jest jednak mechanizm łączący obecność markerów zapalenia we krwi z destabilizacją blaszek miażdżycowych. Słabo poznane są także źródła systemowego podostrego
zapalenia. Możliwych jest kilka scenariuszy:
1. Systemowe zapalenie może być skutkiem a nie przyczyną destabilizacji blaszki; Blaszki
miażdżycowe, w których toczy się aktywny proces zapalny, i które są wobec tego niestabilne, mogą być źródłem albo cytokin prozapalnych stymulujących wątrobową produkcję
CRP, albo/i DAMPs aktywujących odczyn zapalny (ryc. 7.1.);
2. Systemowe zapalenie może być przyczyną destabilizacji blaszki np. poprzez bezpośrednie destabilizujące działanie CRP na blaszki miażdżycowe. CRP jest białkiem ostrej fazy zakażenia/
zapalenia, ale jest także elementem wzmacniającym mechanizm odporności wrodzonej i za174
palenia, dlatego że jest ligandem receptora LOX-1 (rozdz. VII.1). W tym kontekście wykazano
w różnych modelach eksperymentalnych, że CRP wywołuje dysfunkcję śródbłonka naczyniowego, zwiększa ekspresję cząstek adhezyjnych na śródbłonku, zwiększa rekrutację monocytów
do intimy i powstawanie komórek piankowatych, zwiększa naczyniową produkcję ROS i metaloproteinaz oraz zwiększa proliferację i migrację komórek mięśni gładkich [12];
3. Źródłem systemowego zapalenia destabilizującego blaszki miażdżycowe może być, inna
niż miażdżyca, przewlekła choroba zapalna. Dla przykładu, wykazano przeszło dwukrotny
wzrost częstości zawałów i nagłych śmierci sercowych u osób z reumatycznym zapaleniem
stawów [10]. Hipoteza tłumacząca tę statystykę zakłada, że uszkadzane przez proces zapalny stawy są źródłem dużej ilości DAMPs aktywujących wrodzoną odporność i zapalenie
także w lokalizacjach poza stawami [10];
4. Źródłem PAMPs odpowiedzialnych z aktywację wrodzonej odporności i zapalenia może
być normalna flora bakteryjna przewodu pokarmowego, w tym endotoksyna bakteryjna
i LPS, przedostające się w pewnych warunkach do systemowego krążenia [71]. W tym kontekście wykazano, że: (i) bogato-tłuszczowa dieta zwiększa u ludzi i zwierząt przechodzenie
jelitowej endotoksyny i LPS do systemowego krążenia; (ii) źródłem PAMPs stwierdzanych
w krążeniu u ludzi jest przede wszystkim flora bakteryjna obecna w jamie ustnej (vide
związek między chorobami przyzębia i występowaniem miażdżycy), która połykana ze śliną, przechodzi do krążenia głównie na poziomie jelita cienkiego. Odbywa się to w mechanizmie podobnym do wchłaniania witamin rozpuszczalnych w tłuszczach, co polega na
inkorporacji tych substancji do chylomikronów i transporcie z krążeniem limfatycznym do
przewodu piersiowego i krążenia ogólnego; (iii) choć jelito grube jest miejscem o bardzo
bogatej florze bakteryjnej, prawdopodobnie w małym stopniu wpływa na poziom PAMPs
w krążeniu gdyż powierzchna wchłaniania jest w nim bardzo mała a dodatkowo PAMPs
tam wchłaniane trafiają bezpośrednio do wątroby gdzie są eliminowane;
5. Źródłem PAMPs odpowiedzialnych z aktywację wrodzonej odporności i zapalenia może
być infekcja chorobotwórczymi bakteriami lub wirusami; Najczęściej podnoszona jest rolę
zakażeń spowodowanych Chlamydia pneumonie, Helicobacter pylori, Cytomegalovirus oraz
Herpex simplex, które są bardzo częste w populacji ogólnej, i które są często zakażeniami
przewlekłymi przebiegającymi bezobjawowo [72,73]. W zgodzie z tą hipotezą pozostają
obserwacje, że ryzyko OZW jest większe u osób z różnymi zapaleniami (migdałki, górne
drogi oddechowe, pęcherzyk żółciowy). Jak dotąd próba pozytywnej weryfikacji hipotezy infekcyjnej nie powiodła się. Wobec ogromnej powszechności wymienionych zakażeń
i miażdżycy, wyniki badań epidemiologicznych, korelujących poziomy odpowiednich
przeciwciał czy markerów serologicznych z występowaniem miażdżycy, są mało konkluzywne. Dodatkowo badania kliniczne nastawione na eradykację zakażeń bakteryjnych przy
pomocy długotrwałych terapii antybiotykowych nie wykazały by u pacjentów z zawałem,
leczonych antybiotykami, częstość ponownych zawałów malała [74,75];
6. Czynnikiem aktywującym proces zapalny mogą być kwasy tłuszczowe i inne produkty
tłuszczowe, o których wiadomo, że są ligandami receptorów wrodzonej odporności (PRR)
(ryc. VII-2);
7. Czynnikiem aktywującym może być tkanka tłuszczowa, zwłaszcza trzewna, jaka często występuje w nadmiarze u osobników otyłych (rozdz. X).
175
Piśmiennictwo
1. Ross R. Atherosclerosis- an inflammatory disease. N Engl J Med 1999; 340:115-126.
2. Moore KJ, Freeman MW. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler
Thromb Vasc Biol 2006; 26:1702-1711.
3. Cole JE, Georgiou E, Monaco C. The expression and functions of toll-like receptors in atherosclerosis. Mediators Inflamm 2010; 2010:1-18.
4. Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators
Inflamm 2010; Epub@2010 Jul 13.:672395.
5. Shalhoub J, Falck-Hansen MA, Davies AH, Monaco C. Innate immunity and monocyte-macrophage
activation in atherosclerosis. J Inflamm (Lond) 2011; 8:9.
6. Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol
2011; 30:16-34.
7. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like
receptors. Nat Immunol 2010; 11:373-384.
8. Hodgkinson CP, Ye S. Toll-like receptors, their ligands, and atherosclerosis. ScientificWorldJournal
2011; 11:437-53.:437-453.
9. Kennedy DJ, Kashyap SR. Pathogenic Role of Scavenger Receptor CD36 in the Metabolic Syndrome
and Diabetes. Metab Syndr Relat Disord 2011; 9:1-7.
10. Monaco C, Terrando N, Midwood KS. Toll-like receptor signaling: common pathways that drive
cardiovascular disease and rheumatoid arthritis. Arthritis Care Res (Hoboken ) 2011; 63:500-511.
11. Monaco C. The tolls and dangers of atherosclerotic disease. Curr Pharm Biotechnol 2011.
12. Calabro P, Golia E, Yeh ET. CRP and the risk of atherosclerotic events. Semin Immunopathol 2009;
31:79-94.
13. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature 2011; 473:317-325.
14. Miller YI, Choi SH, Wiesner P et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res 2011; 108:235-248.
15. Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-kappaB in the heart: to be or not to
NF-kappaB. Circ Res 2011; 108:1122-1132.
16. Shih HH, Zhang S, Cao W et al. CRP is a novel ligand for the oxidized LDL receptor LOX-1. Am J
Physiol Heart Circ Physiol 2009; 296:H1643-H1650.
17. Fujita Y, Kakino A, Harada-Shiba M et al. C-reactive protein uptake by macrophage cell line via
class-A scavenger receptor. Clin Chem 2010; 56:478-481.
18. Fujita Y, Kakino A, Nishimichi N et al. Oxidized LDL receptor LOX-1 binds to C-reactive protein
and mediates its vascular effects. Clin Chem 2009; 55:285-294.
19. Gill R, Tsung A, Billiar T. Linking oxidative stress to inflammation: Toll-like receptors. Free Radic
Biol Med 2010; 48:1121-1132.
20. Mann DL. The emerging role of innate immunity in the heart and vascular system: for whom the cell
tolls. Circ Res 2011; 108:1133-1145.
21. Greaves DR, Gordon S. The macrophage scavenger receptor at 30 years of age: current knowledge
and future challenges. J Lipid Res 2009; 50 Suppl:S282-S286.
22. Reiss AB, Anwar K, Wirkowski P. Lectin-like oxidized low density lipoprotein receptor 1 (LOX-1) in
atherogenesis: a brief review. Curr Med Chem 2009; 16:2641-2652.
23. Glatz JF, Luiken JJ, Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism:
implications for metabolic disease. Physiol Rev 2010; 90:367-417.
24. Zhang L, Keung W, Samokhvalov V, Wang W, Lopaschuk GD. Role of fatty acid uptake and fatty acid
beta-oxidation in mediating insulin resistance in heart and skeletal muscle. Biochim Biophys Acta
2010; 1801:1-22.
25. Mehta JL, Chen J, Hermonat PL, Romeo F, Novelli G. Lectin-like, oxidized low-density lipoprotein
receptor-1 (LOX-1): a critical player in the development of atherosclerosis and related disorders.
Cardiovasc Res 2006; 69:36-45.
176
26. Puccetti L, Sawamura T, Pasqui AL, Pastorelli M, Auteri A, Bruni F. Atorvastatin reduces plateletoxidized-LDL receptor expression in hypercholesterolaemic patients. Eur J Clin Invest 2005; 35:47-51.
27. Monaco C, Gregan SM, Navin TJ, Foxwell BM, Davies AH, Feldmann M. Toll-like receptor-2 mediates
inflammation and matrix degradation in human atherosclerosis. Circulation 2009; 120:2462-2469.
28. Giannotti G, Landmesser U. Endothelial dysfunction as an early sign of atherosclerosis. Herz 2007;
32:568-572.
29. Landmesser U, Hornig B, Drexler H. Endothelial function: a critical determinant in atherosclerosis?
Circulation 2004; 109:II27-II33.
30. Vanhoutte PM, Shimokawa H, Tang EH, Feletou M. Endothelial dysfunction and vascular disease.
Acta Physiol (Oxf) 2009; 196:193-222.
31. Libby P, Okamoto Y, Rocha VZ, Folco E. Inflammation in atherosclerosis: transition from theory to
practice. CIRC J 2010; 74:213-220.
32. Libby P, Dicarli M, Weissleder R. The vascular biology of atherosclerosis and imaging targets. J Nucl
Med 2010; 51 Suppl 1:33S-37S.
33. Noble MI, Drake-Holland AJ, Vink H. Hypothesis: arterial glycocalyx dysfunction is the first step in
the atherothrombotic process. QJM 2008; 101:513-518.
34. Broekhuizen LN, Mooij HL, Kastelein JJ, Stroes ES, Vink H, Nieuwdorp M. Endothelial glycocalyx
as potential diagnostic and therapeutic target in cardiovascular disease. Curr Opin Lipidol 2009;
20:57-62.
35. Granger DN, Rodrigues SF, Yildirim A, Senchenkova EY. Microvascular responses to cardiovascular
risk factors. Microcirculation 2010; 17:192-205.
36. Caro CG. Discovery of the role of wall shear in atherosclerosis. Arterioscler Thromb Vasc Biol 2009;
29:158-161.
37. Cheng C, Tempel D, van Haperen R et al. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation 2006; 113:2744-2753.
38. Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc
Biol 2007; 27:1248-1258.
39. Worthley SG, Farouque HM, Cameron JD, Meredith IT. Arterial remodeling correlates positively
with serological evidence of inflammation in patients with chronic stable angina pectoris. J Invasive
Cardiol 2006; 18:28-31.
40. Schoenhagen P, Nissen J, Tuzcu M. Coronary arterial remodeling: from bench to bedside. Current
Atherosclerosis Reports 2003; 5:150-154.
41. Hoffmann R, Suliman H, Haager P et al. Association of C-reactive protein and myocardial perfusion
in patients with ST-elevation acute myocardial infarction. Atherosclerosis 2006; 186:177-183.
42. Davies MJ. The birth, growth, and consequences of the atherosclerotic plaque. Dialogs Cardiovasc
Med 1999; 4:115-130.
43. Falk E. Unstable angina with fatal outcome: dynamic coronary thrombosis leading to infarction
and/or sudden death: autopsy evidence of recurrent mural thrombosis with peripheral embolization
culminating in total vascular occlusion. Circulation 1985; 71:699-708.
44. DeWood M, Spores J, Notske R, et al. Prevalence of total coronary occlusion during the early hours
of transmural myocardial infarction. N Engl J Med 1980; 303:897-902.
45. Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation 1995; 92:657-671.
46. Theroux P, Fuster V. Acute coronary syndromes: unstable angina and non-q-wave myocardial infarction. Circulation 1998; 97:1195-1206.
47. Gutstein DE, Fuster V. Pathophysiology and clinical significance of atherosclerotic plaque rupture.
Cardiovasc Res 1999; 41:323-333.
48. Davies MJ. Detecting vulnerable coronary plaques. Lancet 1996; 347:1422-1423.
49. Davies MJ. Stability and instability: two faces of coronary atherosclerosis - the Paul Dudley White
lecture 1995. Circulation 1996; 94:2013-2020.
50. Naghavi M, Libby P, Falk E et al. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part I. Circulation 2003; 108:1664-1672.
177
51. Naghavi M, Libby P, Falk E et al. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part II. Circulation 2003; 108:1772-1778.
52. Fishbein MC. The vulnerable and unstable atherosclerotic plaque. Cardiovasc Pathol 2010; 19:6-11.
53. Rosengren A, Dotevall A, Eriksson H, Wilhelmsen L. Optimal risk factors in the population: prognosis,
prevalence, and secular trends. Data from Göteborg population studies. Eur Heart J 2001; 22:136-144.
54. Alderman EL, Corley SD, Fisher LD et al. CASS Participating Investigatore and Staff. Five-year angiografic follow-up of factors associated with progression of coronary artery disease in the Coronary
Artery Surgery Study (CASS). J Am Coll Cardiol 1993; 22:1141-1154.
55. Yamagishi M, Terashima M, Awano K et al. Morphology of vulnerable coronary plaque: insights
from follow- up of patients examined by intravascular ultrasound before an acute coronary syndrome. J Am Coll Cardiol 2000; 35:106-111.
56. Little WC, Constantinescu M, Applegate RJ et al. Can coronary angiography predict the site of a subsequent myocardial infarction in patients with mild-to-moderate coronary artery disease? Circulation 1988; 78:1157-1166.
57. Danchin N. Is myocardial revascularisation for tight coronary stenoses always necessary? Lancet
1993; 342:224-225.
58. Vaughan CJ, Gotto AM, Basson CT. The evolving role of statins in the management of atherosclerosis. J Am Coll Cardiol 2000; 35:1-10.
59. Roque M, Badimon L, Badimon JJ. Pathophysiology of unstable angina. Thromb Res 1999; 95:V5-V14.
60. Pothula A, Serebruany VL, Gurbel P, Mckenzie ME, Atar D. Pathophysiology and therapeutic modification of thrombin generation in patients with coronary artery disease. Eur J Pharmacol 2000; 402:1-10.
61. Naghavi M, Libby P, Falk E et al. From vulnerable plaque to vulnerable patient. A call for new definitions and risk assessment strategies: Part II. Circulation 2003; 108:1772-1778.
62. Mann JM, Karski JC, Pereira WI, Ramires J, Pileggi F. Histological patterns of atherosclerotic plaques
in unstable angina patients vary according to clinical presentation. Heart 1998; 80:19-22.
63. Gertz SD, Roberts WW. Hemodynamic shear force in rupture of coronary arterial atherosclerosis
plaques. Am J Cardiol 1990; 66:1368-1372.
64. Arroyo LH, Lee RT. Mechanisms of plaque rupture: mechanical and biologic interactions. Cardiovasc Res 1999; 41:369-375.
65. Giles TD. Factors affecting circadian variability. Blood Press Monit 2000; 5 (Suppl 1):S3-S7.
66. Boren J, Gustafsson M, Skalen K, Flood C, Innerarity TL. Role of extracellular retention of low density lipoproteins in atherosclerosis. Curr Opin Lipidol 2000; 11:451-456.
67. Williams KJ, Tabas I. Atherosclerosis; An inflammatory disease. N Engl J Med 1999; 340:1928-1929.
68. Cholesterol Treatment Trialists’ (CTT) Collaboration, Blackwell L, Emberson J, Holland LE et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000
participants in 26 randomised trials. Lancet 2010; 376:1670-1681.
69. Ridker PM, Danielson E, Fonseca FA et al. Rosuvastatin to prevent vascular events in men and
women with elevated C-reactive protein. N Engl J Med 2008; 359:2195-2207.
70. Ridker PM, MacFadyen JG, Fonseca FA et al. Number needed to treat with rosuvastatin to prevent first
cardiovascular events and death among men and women with low low-density lipoprotein cholesterol
and elevated high-sensitivity C-reactive protein: justification for the use of statins in prevention: an intervention trial evaluating rosuvastatin (JUPITER). Circ Cardiovasc Qual Outcomes 2009; 2:616-623.
71. Erridge C. Diet, commensals and the intestine as sources of pathogen-associated molecular patterns in
atherosclerosis, type 2 diabetes and non-alcoholic fatty liver disease. Atherosclerosis 2011; 216:1-6.
72. Muhlestein JB. Chronic infection and coronary artery disease. Med Clin North Am 2000; 84:123.
73. Mehta JL, Romeo F. Inflammation, infection and atherosclerosis - do antibacterials have a role in the
therapy of coronary artery disease? Drugs 2000; 59:159-170.
74. Andraws R, Berger JS, Brown DL. Effects of antibiotic therapy on outcomes of patients with coronary
artery disease: a meta-analysis of randomized controlled trials. JAMA 2005; 293:2641-2647.
75. Anderson JL. Infection, antibiotics, and atherothrombosis--end of the road or new beginnings?
N Engl J Med 2005; 352:1706-1709.
178
VIII. Naczyniowy stres oksydacyjny
Emilia KLEMENSKA
VIII.1. Słowniczek pojęć i terminów wolnorodnikowych
W komórkach organizmów tlenowych, produktem ubocznym różnych enzymatycznych
reakcji oksydoredukcyjnych (vide mitochondrialna fosforylacja oksydacyjna, rozdz. V.8)
jest przeniesienie elektronu na tlen cząsteczkowy (O2) i powstanie cząsteczki tlenu z nadmiarowym, niesparowanym elektronem, która znana jest jako anionorodnik ponadtlenkowy
(O2 + e– O2–).
Atomy, cząsteczki i jony zawierające niesparowany elektron określane są jako wolne rodniki. W tym opracowaniu mowa jest o wolnych rodnikach tlenowych, czyli związkach tlenu
z niesparowanym elektronem. Rodniki tlenowe cechują się dużą reaktywnością chemiczną
(są akceptorem lub donorem elektronu i wobec tego mają właściwości utleniające lub redukujące) i dlatego są nietrwałe, ale i potencjalnie cytotoksyczne. Wiadomo, że obok efektów
toksycznych, ROS pełnią rolę sygnalizacyjną w różnych fizjologicznych procesach regulacyjnych (ryc. 8.1.) [1].
Cytotoksyczność O2– i pochodnych (ryc. 8.1.) wynika z faktu, że substancje te szczególnie
chętnie atakują:
1. tlenek azotu (NO), co ogranicza biodostępność NO i jest przyczyną dysfunkcji śródbłonkowej (rozdz. IX) oraz dodatkowo jest źródłem toksycznego nadtlenoazotynu (O2– + NO
l ONOO–) [2;3];
2. grupy SH białek, co powoduje zmiany właściwości tych białek;
3. lipidy w miejscu ich nienasyconych wiązań, co skutkuje między innymi zaburzeniami struktury fosfolipidów błonowych. Markerem wolnorodnikowej destrukcji fosfolipidów są produkty
ich peroksydacji, malonylodialdehyd i 8-izoprostany, obecne w surowicy i moczu [4];
4. zasady purynowe i pirymidynowe budujące kwasy nukleinowe, co może skutkować zaburzeniami w kodzie genetycznym komórek i mutacjami. Szczególnie podatny na uszkodzenie oksydacyjne jest mitochondrialny DNA. Markerem wolnorodnikowej modyfikacji
kwasów nukleinowych jest 8-hydroksyguanina.
179
Głównymi źródłami ustrojowego O2– są:
1. mitochondria (mitochondrialny łańcuch oddechowy, rozdz. V.8);
2. komórki zapalne, które są wyposażone w enzymy wyspecjalizowane w produkcji bakteriobójczych O2– (leukocytarna oksydaza NADPH) i kwasu podchlorawego (mieloperoksydaza) [5] ;
3. naczynia, w których obecne są przynajmniej trzy enzymy produkujące O2–: naczyniowa
oksydaza NADPH, oksydaza ksantynowa oraz śródbłonkowa syntaza NO (w postaci rozprzęgniętej, rozdz. IX.2.).
O2– i jego toksyczne metabolity są substancjami nietrwałymi i słabo dyfundującymi w środowisku biologicznym, dlatego ich działania, w tym także działania toksyczne, dotyczą bezpośrednio jedynie miejsc gdzie powstają. Z wymienionej listy źródeł ustrojowego O2– wynika,
że toksyczności wolnorodnikowej można oczekiwać zwłaszcza w procesach przebiegających
z (i) zaburzeniami mitochondrialnymi (niedokrwienie/reperfuzja, starzenie, nowotwory); (ii)
aktywacją komórek zapalnych (procesy zapalne i immunologiczne o różnej etiologii i lokalizacji) oraz (iii) w procesach patologicznych z udziałem naczyń (miażdżyca). W kontekście choroby sercowo-naczyniowej, szczególnie dobrze rozpoznany jest udział O2–i jego pochodnych
w uszkodzeniu serca związanym z niedokrwieniem/reperfuzją (w tym, w mechanizmie no-reflow phenomenon) (rozdz. V.10) a także w mechanizmie rozwoju miażdżycy (rozdz. VII).
W naczyniach O2–, przechodzi kaskadę typowych reakcji wolnorodnikowych, w której powstają: nadtlenoazotyn (ONOO–), nadtlenek wodoru (H2O2), rodnik wodorotlenowy (OH*)
i ewentualnie kwas podchlorawy (ryc. 8.1.). Spośród tych substancji, wolnymi rodnikami są
jedynie O2– i OH*. Trzy pozostałe, choć formalnie nie są rodnikami, mają jednak bezpośrednie,
lub za pośrednictwem OH*, liczne działania biologiczne (ryc. 8.1.). Dlatego wszystkie one określane są zbiorczym terminem – reaktywne formy tlenu (ROS, Reactive Oxygen Species).
Komórki organizmów tlenowych wyposażone są w system ochrony anty-rodnikowej, na
który składają się:
1. Enzymy usuwające (dysmutaza ponadtlenkowa) lub zapobiegające powstawaniu ROS (katalaza
i peroksydaza, ryc. 8.1.); Dysmutaza ponadtlenkowa – ma trzy izoformy: Cu/Zn-SOD (SOD-1)
– obecną w cytoplazmie komórek, Mn-SOD (SOD-2) – obecną w mitochondriach, oraz ecSOD
(SOD-3) – zewnątrzkomórkową, związaną z glikokaliksem pokrywającym komórki śródbłonka,
które to enzymy kontrolują poziom O2– tylko we właściwym dla sobie przedziale komórkowym.
Eksperymenty pokazują, że spadkowi aktywności ecSOD towarzyszy wzrost poziomów O2–
i ONOO– w miokardium i dysfunkcja śródbłonkowa. Nadekspresja ecSOD zapobiega dysfunkcji
śródbłonkowej i naczyniowemu odczynowi zapalnemu w modelu cukrzycy. U pacjentów z cukrzycą stwierdzono zmniejszone wiązanie ecSOD z powierzchnią komórek śródbłonka i objawy
stresu oksydacyjnego. Podanie rekombinowanej ecSOD pacjentom z nadciśnieniem skutkowało
obniżeniem ciśnienia. Razem wziąwszy, wszystko przemawia za hipotezą, że to ecSOD jest strażnikiem poziomu ROS w przestrzeni międzykomórkowej i świetle naczyń [6].
2. Nieenzymatyczne wymiatacze wolnych rodników – kilka drobnocząsteczkowych substancji obecnych w cytoplazmie komórek i/lub błonach komórkowych (np. β-karoten,
α-tokoferol, witamina C, koenzym Q10, ubichinon, selen, melatonina), które, ze względu
na swe właściwości chemiczne, są w sposób preferencyjny atakowane i degradowane przez
ROS, dzięki czemu zapobiegają wolnorodnikowemu uszkodzeniu innych życiowo ważnych
składników komórkowych.
180
Ryc. 8.1. Reakcje z udziałem O2– w naczyniach. Reakcja O2– z NO jest preferowana gdyż jest najszybsza
(6,7 x 109 mol/L/sec). Obok reakcji z NO, zachodzą dwie reakcje dysmutacji O2– do H2O2: szybka – przy
udziale dysmutazy ponadtlenkowej (SOD) (1,8 x 109 mol/L/sec) oraz ~10 tys. razy wolniejsza – spontaniczna, bez udziału SOD (8,0 x 104 mol/L/sec). Produktem rozpadu nadtlenoazotynu i nadtlenku
wodoru może być toksyczny rodnik hydroksylowy (OH*). Zwykle jednak H2O2 jest rozkładany do
wody (bez udziału OH*) przez enzymy katalazę i peroksydazę glutationu. H2O2 przekształca się w OH*
jedynie w obecności jonów zredukowanego żelaza (Fe2+) lub miedzi (Cu+) (reakcja Fentona) i w tym
mechanizmie metale te mogą być źródłem toksyczności wolnorodnikowej. W obecności mieloperoksydazy uwalnianej z aktywowanych komórek zapalnych, H2O2 przekształca się w kwas podchlorawy (kwas
chlorowy I) – silny oksydant o działaniu bakteriobójczym. O2– i jego pochodne ROS, wszystkie mają
liczne działania biologiczne, częściowo regulacyjne, a częściowo toksyczne.
W różnych stanach patologicznych, w tym pod wpływem czynników ryzyka sercowonaczyniowego, dochodzi do zaburzenia równowagi między produkcją i eliminacją ROS. Towarzyszy temu wzrost lokalnego stężenia ROS i objawy ich toksycznego działania na tkanki.
Stan taki określany jest terminem stres oksydacyjny.
VIII.2. ROS w naczyniach
NO jest głównym mediatorem śródbłonkowym o działaniu przeciwzapalnym i przeciwmiażdżycowym. Bodźcami pro-zapalnymi są śródbłonkowy niedobór NO oraz nadmiar O2–
i innych ROS. Główną przyczyną niedoboru śródbłonkowego NO i rozwoju naczyniowego
odczynu zapalnego, jaki leży u podstawy: (i) mechanizmu miażdżycy; (ii) destabilizacji już
ukształtowanych blaszek miażdżycowych oraz (iii) zjawiska no-reflow phenomenon, typowego dla reperfundowanych organów (rozdz. V.10) jest naczyniowa nadprodukcja O2– i innych
ROS (ryc. 8.2.).
181
Ryc. 8.2. Udział stresu oksydacyjnego w rozwoju pro-zapalnego fenotypu śródbłonka naczyniowego.
Biologiczna dostępność O2– i innych ROS i ich efekty biologiczne są wypadkową równoczesnej produkcji i degradacji ROS. Zewnątrzkomórkowa dysmutaza ponadtlenkowa (ecSOD), związana z glikokaliksem, jest głównym strażnikiem wewnątrznaczyniowego stężenia O2– i pośrednio innych ROS. Wiązanie ecSOD z uszkodzonym glikokaliksem maleje i naczyniowy poziom ROS rośnie (prawa strona).
Komórki zapalne przylegające do powierzchni śródbłonka produkują ROS, co wzmacnia pro-zapalną
stymulację śródbłonka (lewa strona).
Rolę „dyrygenta” naczyniowego stresu oksydacyjnego pełni prawdopodobnie naczyniowa oksydaza NADPH, której aktywność jest regulowana przez różne czynniki humoralne
i hormonalne i która, z kolei, jest regulatorem aktywności wolnorodnikowej oksydazy ksantynowej i śródbłonkowej syntazy NO (eNOS) (ryc. 8.3.). W ten sposób produkcja O2– inicjowana przez oksydazę NADPH jest prawdopodobnie wzmacniana przez dwa pozostałe enzymy.
Wtórnie do zwiększonej naczyniowej produkcji O2– maleje biodostępność NO. NO jest czynnikiem hamującym aktywność oksydazy NADPH. Dlatego wszystkie czynniki ograniczające
biodostępność NO mogą być powodem zwiększonej naczyniowej produkcji O2– i stresu oksydacyjnego. Innymi słowy, stres oksydacyjny może być źródłem dysfunkcji śródbłonkowej a ta,
na drodze sprzężenia zwrotnego, źródłem stresu oksydacyjnego. Rozumowanie to stanowiło
podstawę klinicznych prób modyfikowania przebiegu choroby sercowo-naczyniowej przy pomocy leków o działaniu antyrodnikowym (rozdz. VIII.4).
Oksydaza NADPH; Długo sądzono, że enzym ten występuje jedynie w leukocytach,
gdzie odpowiedzialny jest za produkcję bakteriobójczego O2–. Obecnie wiadomo, że oksydaza
NADPH (substratem do produkcji O2– jest NADPH i O2) o zbliżonej strukturze i właściwościach do enzymu leukocytarnego, występuje także w komórkach śródbłonka i komórkach
naczyniowych mięśni gładkich. Enzym jest pięcio-białkowym kompleksem, którego jednostka katalityczna (gp91phox) jest zakotwiczona w błonie komórkowej i wobec tego uwalnia
182
Ryc. 8.3. Schemat prawdopodobnych związków między naczyniowymi enzymami produkującymi O2–.
O2– produkowany przez oksydazę NADPH powoduje przekształcenie dehydrogenazy ksantynowej
w oksydazę, który to enzym produkuje O2–. W wyniku interakcji O2– z NO powstaje nadtlenoazotyn,
który jest substancją degradującą tetrahydrobiopterynę (BH4). Pod nieobecność BH4, syntaza NO,
obok NO produkuje także O2–. NO jest inhibitorem oksydazy NADPH. Mała biodostępność NO skutkuje odblokowaniem (aktywacją) tego enzymu.
O2– częściowo do cytoplazmy a częściowo do przestrzeni pozakomórkowej. W przeciwieństwie do enzymu leukocytarnego, naczyniowa oksydaza NADPH konstytutywnie produkuje
pewne ilości O2– i jej aktywność i ekspresja są dodatkowo zwiększane przez takie czynniki
jak: angiotensyna, endotelina, aldosteron, cytokiny pro-zapalne (TNF-α, TGF-β) [7] i inne
[8,9]. Natomiast NO zmniejsza aktywność oksydazy NADPH. Istnieje coraz więcej dowodów
na to, że dysfunkcja śródbłonkowa towarzysząca hipercholesterolemii, cukrzycy, nadciśnieniu tętniczemu, miażdżycy, niewydolności serca czy tolerancji azotanów jest spowodowana
zwiększoną produkcją O2– przez naczyniową oksydazę NADPH. Badania eksperymentalne
dowodzą także, że eliminacja oksydazy NADPH (poprzez knock-out genu), zapobiega rozwojowi miażdżycy w różnych modelach eksperymentalnych.
Oksydoreduktaza ksantynowa; Jest enzymem zewnątrzkomórkowym, związanym z glikokaliksem, przekształcającym hipoksantynę i ksantynę (produkty katabolizmu nukleotydów
adeninowych, ryc. 5.3) w kwas moczowy i blokowanym przez allopurinol. W normalnych warunkach enzym istnieje w formie dehydrogenazy ksantynowej. Natomiast w wyniku działania
na enzym ROS pochodzących z NADPH oksydazy, przekształca się on formę oksydazy ksantynowej, która obok kwasu moczowego produkuje dodatkowo O2–. Obok NADPH oksydazy
regulatorem oksydazy ksantynowej są także niedotlenienie, cytokiny prozapalne i lipopolisacharydy. Wzrost aktywności oksydazy ksantynowej towarzyszy także niedokrwieniu/reperfuzji narządów, chorobie wieńcowej i niewydolności serca [10]. Na udział oksydazy ksantynowej
183
w chorobach układu krążenia wskazują korzystne efekty kliniczne allopurinolu. Allopurinol
poprawił funkcję śródbłonka w prewencji pierwotnej u pacjentów wysokiego ryzyka z zespołem
metabolicznym i cukrzycą typu 2 lub nadciśnieniem i towarzyszył temu spadek osoczowego
poziomu malonyldialdehydu (MDA, marker stresu oksydacyjnego). Inne prace donosiły o poprawie funkcji śródbłonka i spadku poziomu MDA a także o zwiększonej tolerancji wysiłku
w teście wysiłkowym u pacjentów ze stabilną chorobą wieńcową leczonych allopurinolem [11].
VIII.3. Rola wolnorodnikowej modyfikacji LDL w patomechanizmie
miażdżycy
Główne argumenty na rzecz udziału wolnorodnikowej modyfikacji LDL w patomechanizmie miażdżycy są następujące [12]:
1. Monocyty/makrofagi w hodowli komórkowej znacznie szybciej obładowują się ox-LDL niż
natywną formą LDL;
2. ox-LDL, działając głównie za pośrednictwem receptora LOX-1, indukują potencjalnie proaterogenne efekty, w tym: (i) są cytotoksyczne dla śródbłonka (dane z hodowli komórkowej); (ii) stymuluja komórki śródbłonka do produkcji MCP-1 (chemoatraktant nasilający
adhezję monocytów); (iii) nasilają produkcję kolagenu w komórkach mięśniówki gładkiej;
(iv) nasilają apoptozę; (v) hamują uwalnianie NO i zwiększają ekspresję VCAM-1 w komórkach śródbłonka; (vi) zwiększają ekspresję czynnika tkankowego oraz (vii) indukują
szereg cytokin prozapalnych w makrofagach;
3. Rozwój miażdżycy w modelach eksperymentalnych jest spowolniony przez interwencje
antyrodnikowe.
VIII.4. Próby kliniczne z interwencjami anty-rodnikowymi
Większość dużych badań klinicznych z antyoksydantami testowała efekty witaminy E
oraz β-karotenu. Metanaliza dużych badań klinicznych (n = ~80 tys. pacjentów, dawki
50–400 mg/d) dotyczących populacji generalnej pacjentów z chorobą wieńcową, cukrzyca
czy nadciśnieniem nie wykazała by stosowanie tych leków miało korzystny wpływ na częstość
występowania wydarzeń sercowo-naczyniowych [13].
Przyczyna niepowodzeń klinicznych z antyoksydantami jest zastanawiająca, zwłaszcza
wobec faktów, że antyoksydanty zapobiegają różnym eksperymentalnym postaciom miażdżycy. Podnoszone są następujące możliwe przyczyny niepowodzeń:
1. Witamina E może być nieodpowiednim antyoksydantem u ludzi, używane dawki były zbyt
małe, lek był stosowany w zbyt późnym stadium rozwoju miażdżycy, lub może jest skuteczny tylko w niektórych podgrupach pacjentów (vs. populacja ogólna). W tym ostatnim
kontekście przytacza się korzystne efekty witaminy E w podgrupach pacjentów o szczególnie nasilonym stresie oksydacyjnym. W badaniu SPACE, dotyczącym 196 dializowanych pacjentów z niewydolnością nerek randomizowanych do grupy kontrolnej lub grupy
z witaminą E w wysokiej dawce (800 mg/d) obserwowano >50% redukcję występowania
wydarzeń sercowo-naczyniowych [14]. Podobną 47% redukcję wydarzeń obserwowano
w grupie 1434 pacjentów z cukrzycą i genotypem haptoglobina 2–2 (grupa o szczególnie
184
nasilonym stresie oksydacyjnym i 2–5-krotnie zwiększonym ryzyku wydarzeń sercowo naczyniowych) leczonych witaminą E (400 mg/d) [15].
2. Antyoksydanty/wymiatacze ROS są nieskuteczne klinicznie gdyż, w istocie, przeważa ich
działanie likwidujące korzystne biologicznie efekty ROS. Wiadomo, że w niewielkich stężeniach ROS pełnią ważne funkcje sygnalizacyjne;
3. Krańcowo odmienna hipoteza jest taka, że antyoksydanty nie mogą mieć korzystnego działania gdyż nie mają szansy skutecznie przeciwdziałać stresowi oksydacyjnemu ze względu
na zbyt małe ich stężenia osiągane w wyniku suplementacji. Chodzi o to, że reakcja antyoksydant-ROS jest o kilka rzędów wielkości wolniejsza od reakcji O2–-NO, O2–-SOD, czy
reakcji spontanicznej dysmutacji O2–. Dlatego jest mało prawdopodobne by antyoksydant
mógł konkurować z tymi szybkimi i toksycznymi reakcjami. Prawdopodobnie skuteczniejszą strategią mogłoby być hamowanie reakcji syntezy ROS, najlepiej jedynie w strategicznie
ważnych lokalizacjach, np. w ścianie naczyniowej.
4. Nie można całkowicie wykluczyć możliwości, że hipoteza o udziale ROS w patofizjologii
choroby sercowo-naczyniowej jest błędna.
Piśmiennictwo
1. Droge W. Free radicals in the physiological control of cell function. Physiol Rev 2002; 82:47-95.
2. Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to
menace. Circulation 2006; 113:1708-1714.
3. Forstermann U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch 2010; 459:923-939.
4. Lawson JA, Rokach J, Fitzgerald GA. Isoprostanes: formation, analysis and use as indices of lipid
peroxidation in vivo. J Biol Chem 1999; 274:24441-24444.
5. Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch Biochem Biophys 2002;
397:342-344.
6. Fattman CL, Schaefer LM, Oury TD. Extracellular superoxide dismutase in biology and medicine.
Free Radic Biol Med 2003; 35:236-256.
7. Rueckschloss U, Duerrschmidt N, Morawietz H. NADPH oxidase in endothelial cells: impact on
atherosclerosis. Antioxid Redox Signal 2003; 5(2):171-180.
8. Lassegue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 2010; 30:653-661.
9. Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase. Role in Cardiovascular biology and
disease. Circ Res 2000; 86:494-501.
10. Berry CE, Hare JM. Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms
and pathophysiological implications. J Physiol 2004; 555:589-606.
11. Antony R, Dargie HJ. Allopurinol for chronic stable angina: old drug, new tricks? Lancet 2010;
375:2126-2127.
12. Steinberg D. The LDL modification hypothesis of atherogenesis: an update. J Lipid Res 2009; 50
(Suppl):S376-S381.
13. Kris-Etherton PM, Lichtenstein AH, Howard BV, Steinberg D, Witztum JL. Antioxidant vitamin
supplements and cardiovascular disease. Circulation 2004; 110:637-641.
14. Boaz M, Smetana S, Weinstein T et al. Secondary prevention with antioxidants of cardiovascular disease in endstage renal disease (SPACE): randomised placebo-controlled trial. Lancet 2000;
356:1213-1218.
15. Blum S, Vardi M, Brown JB et al. Vitamin E reduces cardiovascular disease in individuals with diabetes mellitus and the haptoglobin 2-2 genotype. Pharmacogenomics 2010; 11:675-684.
185
IX. Śródbłonek naczyniowy
i jego dysfunkcja
Michał MĄCZEWSKI, Anna KONIOR
IX.1. Budowa i role biologiczne śródbłonka
Wnętrze naczyń krwionośnych i limfatycznych jest wysłane jedną warstwą ściśle do siebie przylegających komórek śródbłonka, przytwierdzonych integrynowymi cząstkami adhezyjnymi do błony podstawnej i macierzy zewnątrzkomórkowej. Komórki te komunikują się
z komórkami naczyniowych mięśni gładkich przy pomocy połączeń szczelinowych (gap junctions), przez które mogą przepływać z komórki do komórki: prąd elektryczny, jony i drobnocząsteczkowe substancje. Od strony światła naczynia śródbłonek jest pokryty ~500 nm
warstwą glikokaliksu zbudowanego z proteoglikanów i glikozaminoglikanów z dominacją
siarczanu heparanu.
Śródbłonek naczyniowy człowieka ma dużą całkowitą masę (~1 kg, ~6 x 1013 komórek)
i powierzchnię (~700 m2) [27] i stanowi szczelną barierę odgradzającą krew, w tym białka układu krzepnięcia, płytki i komórki zapalne, od tkanki łącznej i warstwy mięśniowej ściany naczyniowej. Dzięki dużej masie, strategicznej lokalizacji, faktowi, że ma zdolność do odbierania
bodźców ze środowiska zewnętrznego (bodźce hemodynamiczne, pO2, bodźce chemiczne),
oraz reagowania na te bodźce zmianą produkcji licznych substancji czynnych (tab. 9.1.) i ekspresji białek, śródbłonek jest organem uczestniczącym w utrzymywaniu homeostazy układu
krążenia, a zwłaszcza jest regulatorem [7]:
1. skurczu i rozkurczu mięśni gładkich budujących ścianę naczyniową (rozdz. III);
2. rozrostu mięśni gładkich ściany naczyniowej, co razem z regulacją ich napięcia decyduje
o oporze naczyniowym i lokalnym przepływie krwi;
3. ekspresji cząstek adhezyjnych z rodziny selektyn (selektyna P i E) oraz adresyn immunoglobulinopochodnych (VCAM-1, ICAM-1, ICAM-2, PECAM-1) oraz czynników chemotaktycznych komórek zapalnych (MCP-1), które umożliwiają adhezję komórek zapalnych
i płytek do powierzchni śródbłonka, ich aktywację oraz migrację komórek zapalnych do
187
Tabela 9.1. Substancje czynne produkowane przez śródbłonek naczyniowy i ich efekty działania
Substancja
Tlenek azotu
Prostacyklina
Tk. aktywator
plazminogenu (tPA)
Bradykinina
Czynnik stymulujący uwalnianie
Siły hydrodynamiczne: siła
ścinająca, napięcie pulsacyjne;
Substancje uwalniane z płytek:
ADP, ATP, serotonina, trombina,
czynnik aktywujący płytki (PAF);
Acetylocholina, bradykinina,
substancja P, noradrenalina,
histamina, wazopresyna, endotelina;
Siła ścinająca, ADP, ATP,
serotonina, niedotlenienie;
Podobne czynniki jak NO;
Efekt
napięcie i proliferacja
m. gładkich naczyń;
adhezja i agregacja płytek,
dezagregacja agregatów
płytkowych;
adhezja granulocytów
i monocytów do śródbłonka;
produkcja endoteliny;
adhezja i agregacja płytek;
proliferacja m. gładkich;
fibrynoliza
Podobne czynniki jak NO;
napięcie i proliferacja
m. gładkich naczyń;
napięcie m. gładkich naczyń;
Śródbłonkowy czynnik Podobne czynniki jak NO;
hyperpolaryzujący
(EDHF)
Endotelina-1
Siła ścinająca, trombina,
angiotensyna II, adrenalina,
wazopresyna, endotelina,
niedotlenienie, niedobór NO;
Tromboksan A2
Prostaglandyna H2
Angiotensyna II, acetylocholina,
serotonina, histamina;
Angiotensyna II
Niedokrwienie, niedotlenienie;
Anionorodnik (O2– )
Angiotensyna II,
hypercholesterolemia, cukrzyca,
otyłość, inne
napięcie i proliferacja
m. gładkich;
produkcja NO (niskie stężenia
ET-1);
produkcja O2–
napięcie i proliferacja
m. gładkich;
Antagonizują efekty NO
i prostacykliny;
napięcie i proliferacja
m. gładkich;
śródbłonkowa produkcja O2– ;
produkcja PAI;
Degradacja NO i liczne
konsekwencje braku NO;
intimy, co razem wziąwszy określane jest jako proces zapalny w ścianie naczyniowej i co
ewentualnie skutkuje rozwojem zmian miażdżycowych;
4. adhezji i agregacji płytek krwi;
5. procesów krzepnięcia i fibrynolizy.
Niektóre z mediatorów śródbłonkowych działają hamująco (NO, prostacyklina), a inne
stymulująco na każdy z wymienionych procesów (endotelina, angiotensyna) (tab. 9.1.). Ostateczny efekt działania śródbłonka ustala się w wyniku sumowania się tych przeciwstawnych
wpływów. W naczyniach ze zdrowym śródbłonkiem przeważa jego działanie rozkurczające i antyproliferacyjne na mięśniówkę ściany naczyniowej, antypłytkowe, antykoagulacyjne
i profibrynolityczne oraz przeciwzapalne. Czynnikom ryzyka choroby sercowo-naczyniowej
188
towarzyszy zwykle dysfunkcja śródbłonkowa i wtedy efektem jego działania są: (i) wzrost
napięcia i przebudowa ściany naczyniowej; (ii) naczyniowy odczyn zapalny oraz (iii) zwiększona adhezja i agregacja płytek i wzrost krzepliwości a więc trzy podstawowe procesy leżące
u podstawy mechanizmu miażdżycy i destabilizacji już ukształtowanych blaszek miażdżycowych (tzw. promiażdżycowy fenotyp śródbłonka).
IX.2. Tlenek azotu – główny mediator śródbłonkowy
NO jest głównym mediatorem śródbłonkowym i jego niedobór jest ważną przyczyną rozwoju promiażdżycowego fenotypu śródbłonka.
Głównym źródłem NO w naczyniach jest śródbłonkowa izoforma syntazy NO (eNOS)
ale w układzie sercowo-naczyniowym obecne są także izoformy: neuronalna (nNOS) i indukowalna (iNOS). Aktualna dostępność biologiczna NO jest wypadkową produkcji NO i stale
odbywającej się jego inaktywacji przez O2– (ryc. 9.1.).
Ryc. 9.1. Biologiczna dostępność NO i jego efekty biologiczne jako wypadkowa równoczesnej produkcji i degradacji NO. Wielkość produkcji NO jest regulowana głównie przez laminarny przepływ krwi. Czynnikiem
degradującym NO jest O2– stale produkowany nawet w normalnych naczyniach. Czynniki ryzyka choroby
sercowo-naczyniowej zwiększają naczyniową produkcje O2–, co skutkuje mniejszą biodostępnością NO.
Podstawowym czynnikiem podtrzymującym śródbłonkową produkcję NO jest przepływ
laminarny krwi w naczyniu. Natomiast wielkość naczyniowej produkcji O2– jest warunkowana obecnością i nasileniem czynników ryzyka (nawet w normalnym naczyniu ma miejsce
niewielka toniczna produkcja O2–). Reakcja NO + O2– ONOO–: (i) zachodzi bardzo szybko
gdyż jest limitowana jedynie szybkością dyfuzji NO i O2–; (ii) jest głównie odpowiedzialna
(obok hemoglobiny) za unieczynnianie naczyniowego NO oraz (iii) jest źródłem potencjalnie
toksycznego nadtlenoazotynu (vide poniżej).
Cząsteczki eNOS są nieaktywne, kiedy są związane, poprzez błonowe białko kaweolinę,
z wewnętrzną powierzchnią błony komórkowej śródbłonków. Odłączenie eNOS od kaweoliny aktywuje enzym. Znane są przynajmniej trzy mechanizmy aktywacji eNOS [8]:
189
1. Najważniejsza jest aktywacja w wyniku działania laminarnej siły ścinającej; Wzrost przepływu krwi i siły ścinającej (np. pod wpływem wysiłku, SS = 4Qη/Πr3) (rozdz. III.2.3)
powoduje odkształcenie glikokaliksu i integryn na powierzchni komórek śródbłonka, co
skutkuje kolejno: aktywacją kinazy tyrozynowej, fosforylacją eNOS przez kinazę, odłączeniem eNOS od kaweoliny i aktywacją enzymu w mechanizmie niezależnym od Ca2+ (ryc.
9.2.A, prawa strona). Przepływ laminarny i siła ścinająca z jednej strony podtrzymują podstawowe wytwarzanie NO przez śródbłonek naczyń tętniczych i żylnych, a z drugiej zapobiegają inaktywacji NO przez O2–. Chodzi o to, że przepływ laminarny zwiększa ekspresję
zewnątrzkomórkowej dysmutazy ponadtlenkowej (ecSOD), enzymu który jest związany
z glikokaliksem śródbłonkowym i który unieczynnia O2–. Przepływ burzliwy, objawiający
się różnymi zawirowaniami przepływu i zmianami jego kierunku nie tylko nie podtrzymuje tonicznego wydzielania NO, ale jest czynnikiem stymulującym śródbłonkową produkcję
O2–, apoptozę komórek śródbłonka i rozwój miażdżycy, [10] aktywując NOS poprzez odłączanie enzymu od kaweoliny;
2. Przedłużające się działanie siły ścinającej, związane np. z treningiem fizycznym, skutkuje
dodatkowo wzrostem ekspresji eNOS, co jest związane z wzrostem częściowo transkrypcji
(poprzez aktywację NF-κB) (ryc. 9.2.A, lewa strona) a częściowo stabilności mRNA dla
eNOS. Zjawisko to jest także przyczyną tzw. arterializacji śródbłonka naczyniowego w żylnych pomostach naczyniowych [29].
(A)
(B)
Ryc. 9.2. Wpływ laminarnego przepływu na aktywność i ekspresję eNOS i ekspresje genów prozapalnych w naczyniach z prawidłowym i dysfunkcjonalnym śródbłonkiem. (A) – w prawidłowym śródbłonku przepływ laminarny zwiększa zarówno aktywność jak i częściowo ekspresję eNOS (wynik zwiększonej ekspresji i stabilności mRNA dla eNOS). Stymuluje także ekspresję genów prozapalnych. Zmiany
w ekspresji genów są jednak ograniczane przez NO, którego produkcja rośnie i który hamuje aktywność
NF-κB. (B) – w obecności czynników zmniejszających biodostępność i/lub produkcję NO laminarny
przepływ zwiększa ekspresję genów prozapalnych, gdyż nie są one zwrotnie hamowana przez NO.
190
Tabela 9.2. Substancje czynne i ich receptory aktywujące wytwarzanie NO przez śródbłonek.
Substancja
Acetylocholina
Bradykinina
Serotonina
Noradrenalina
Histamina
Endotelina
Angiotensyna II
Substancja P
Receptor
Muskarynowy M2
B2
5-HT
α1-adrenergiczny; β2-adrenergiczny
H2
ET-B
AT2
3. Aktywacja w wyniku stymulacji receptorów błonowych komórek śródbłonka przez substancje czynne (tab. 9.2.). Jej efektem jest wzrost komórkowego stężenia Ca2+ i wypieranie
eNOS z połączenia z kaweoliną przez kompleks Ca2+-kalmodulina.
eNOS jest oksydoreduktazą o budowie homodimeru (ryc. IX-3). Enzymatyczny mechanizm syntezy NO przez eNOS polega na utlenianiu aminokwasu L-argininy do L-cytruliny
w obecności kofaktorów: NADPH, nukleotydów flawinowych FAD i FMN oraz hemu i tetrahydrobiopteryny (BH4):
L-arginina + NADPH + O2 → L-cytrulina + NADP + H2O + NO
q
FAD, FMN, hem, BH4
Ryc. 9.3. Schemat homodimeru eNOS i przepływu elektronów pomiędzy jego składowymi monomerami.
Kierunek przepływu elektronów pokazują strzałki; Fe – żelazo w hemowej grupie prostetycznej eNOS.
BH4, tetrahydrobiopteryna związana z hemową grupa prostetyczną enzymu. Arg – L-arginina. wg. [8].
W każdym monomerze enzymu odbywa się wędrówka elektronu z O2 NADPH FAD FMN. Następnie elektron przechodzi do grupy prostetycznej enzymu na bliźniaczym
monomerze, gdzie w obecności BH4, następuje przeniesienie elektronu na azot reszty guanidynowej L-argininy, uwolnienie NO i powstanie L-cytruliny. W sytuacjach niedoboru L-argininy albo BH4, przechodzenie elektronu na sąsiedni monomer jest niemożliwe (następuje
tzw. rozprzęganie eNOS) i zamiast na L-argininę, elektron jest przenoszony na O2, czemu
towarzyszy powstawanie O2–. Nawet w normie eNOS produkuje, obok NO, niewielkie ilości
191
O2–. Proporcje te zmieniają się na korzyść O2– w wyniku rozprzęgania eNOS [8]. Rozprzęganie jest stanem sprzyjającym postawaniu toksycznego nadtlenoazotynu, który jest substancją
silnie utleniającą, między innymi BH4.
NO jest lipofilną substancją łatwo penetrującą przez błony komórkowe i aktywnie reagującą z O2– O2 i białkami zawierającymi w swojej strukturze hem. Z tego powodu okres
półtrwania NO w środowisku naczyniowym jest bardzo krótki (2–3 sek), co sprawia, że jest
substancją o działaniu głównie lokalnym.
NO reagując z O2– i O2 ulega utlenianiu, odpowiednio, do toksycznego nadtlenoazotynu oraz
azotynów (NO2–). W wyniku interakcji z białkami zawierającymi hem (np. hemoglobina), NO ulega utlenieniu do azotanów (NO3–), które są głównym stabilnym metabolitem NO w płynach ustrojowych. NO aktywuje niektóre białka hemowe (cyklaza guanylowa) a inne hamuje (cytochrom
P450, katalaza, oksydaza cytochromowa C), powoduje także powstanie methemoglobiny [8].
Komórkowe mechanizmy odpowiedzialne za fizjologiczne efekty śródbłonkowego NO to:
1. Stymulacja przez NO cytoplazmatycznej rozpuszczalnej cyklazy guanylowej, zwiększona
produkcja przez ten enzym cGMP i aktywacja przez cGMP zarówno: (i) kinazy białkowej G i fosforylacji przez tę kinazę różnych białek efektorowych jak i (ii) fosfodiesterazy II
rozkładającej cAMP i zmniejszającej fosforylację związaną z systemem cAMP-kinaza białkowa A (aktywowanym głównie przez stymulacji receptorów adrenergicznych β). Z działaniem via cGMP związane są efekty NO: naczynio-rozszerzające, przeciwpłytkowe NO
i anty-proliferacyjne w stosunku do naczyniowych mięśni gładkich [8];
2. Bezpośrednie hamowanie mitochondrialnej oksydazy cytochromowej C i hamowanie oddychania mitochondrialnego [8];
3. Działanie poprzez produkt reakcji NO + O2– ONOO–; W obecności krótkotrwałej nadprodukcji O2– wynikiem tej reakcji jest detoksyfikacja O2– kosztem zmniejszonej biodostępności NO. W obecności dłużej trwającego stresu oksydacyjnego, dominującym efektem
powstawania nadtlenoazotynu jest utlenianie BH4, rozprzęganie eNOS, zmniejszona produkcja przez enzym NO i zwiększona O2– i jeszcze większa nadprodukcja nadtlenoazotynu.
Cukrzyca jest przykładem patologii, w której rozprzęganie eNOS uczestniczy w mechanizmie naczyniowego stresu oksydacyjnego i dysfunkcji śródbłonkowej [4];
4. Nitrozylacja przez NO reszt cysteinowych różnych białek. W tym mechanizmie, w zdrowym śródbłonku, przepływ laminarny i NO inaktywują czynnik transkrypcyjny NF-κB,
co skutkuje mniejszą ekspresją genów: (i) cząstek adhezyjnych (selektyny E i P, ICAM-1,
VCAM-1); (ii) chemokin o działaniu chemotaktycznym w stosunku do komórek zapalnych (np. MCP-1) oraz (iii) prokrzepliwego czynnika tkankowego (ryc. 9.2.A, lewa strona).
Ten sam przepływ laminarny ma działanie prozapalne i prokrzepliwe w stanach, w których
biodostępność śródbłonkowego NO jest upośledzona (np. stres oksydacyjny, rozprzęganie eNOS, niedobór L-argininy i BH4, polimorfizm promotora eNOS, nadprodukcja endogennego inhibitora eNOS) i wobec tego nie ma zwrotnego hamowania NF-κB przez
NO (ryc. 9.2.B, lewa strona). Główną przyczyną niedoboru śródbłonkowego NO jest stres
oksydacyjny. ROS, same przez się, są czynnikiem aktywującym NF-κB i ekspresję prozapalnych genów [8]. Stąd aktywacja prozapalnego fenotypu śródbłonka jest szczególnie silna
w obecności stresu oksydacyjnego gdyż jest wynikiem sumowania się efektów nadmiaru
ROS i niedoboru NO.
192
IX.3. Regeneracja śródbłonka
Komórki śródbłonka, jak wszystkie komórki nabłonkowe, żyją krótko, kilka dni. Systematycznie ulegają oddzielaniu od błony podstawnej i złuszczaniu do krwi. Tak powstają tak zwane krążące komórki śródbłonka (circulating endothelial cells, CEC). Komórki śródbłonka mogą
także rozpadać się na mniejsze fragmenty, tworząc tzw. śródbłonkowe mikrocząstki (endothelial microparticles). Oba te procesy ulegają przyspieszeniu pod wpływem znanych czynników
ryzyka dysfunkcji śródbłonka (hipercholesterolemii, cukrzycy, palenia papierosów).
W regeneracji śródbłonka uczestniczą dwa mechanizmy: (i) miejscowe mnożenie się komórek śródbłonka przez podział oraz (ii) regeneracja z udziałem śródbłonkowych komórek
macierzystych (endothelial progenitor cells, EPC) importowanych do miejsca ubytku śródbłonkowego ze szpiku kostnego. Osiedlają się w miejscach wysokiego stężenia lokalnych czynników (np. SDF-1). Mobilizacja ich jest częściowo NO-zależna (zwiększa ją trening fizyczny),
obniżona w obecności typowych czynników ryzyka dysfunkcji śródbłonkowej, poprawiana
przez wysiłek fizyczny i statyny. Czyli czynniki ryzyka miażdżycy, np. hipercholesterolemia
przesuwają równowagę starzenie się/odnowa śródbłonka na rzecz tej pierwszej.
IX.4. Naczynio-rozkurczające i przeciwmiażdżycowe działanie śródbłonka
Laminarny przepływ krwi, skutkujący zwiększoną aktywnością i/lub ekspresją eNOS jest
podstawowym elementem śródbłonkowej regulacji lokalnego przepływu krwi oraz przeciwzapalnego, przeciwkrzepliwego i profibrynoliticznego (w sumie antymiażdżycowego) działania śródbłonka. Laminarny przepływ i właściwa aktywność eNOS warunkują dodatkowo
sprawną regenerację śródbłonka (rozdz. IX.3.). Paradoksalnie jednak, ten sam laminarny
przepływ i związana z nim siła ścinająca mogą w obszarach naczyniowych z dysfunkcją śródbłonka skutkować upośledzeniem lokalnego przepływu i rozwojem promiażdżycowego fenotypu śródbłonka (ryc. 9.2.).
IX.4.1. Śródbłonkowa regulacja przepływu tkankowego
Błona mięśniowa naczyń znajduje się pod stałym działaniem czynników kurczących
i rozkurczających. Aktualne napięcie ściany naczyniowej (tonus) jest wynikiem sumowania się
tych efektów. Za część skurczową tonusu odpowiada regulacja miogenna, a za część rozkurczową – regulacje metaboliczna i śródbłonkowa związana z uwalnianiem NO (rozdz. III.3).
Dysfunkcja śródbłonkowa, skutkuje trwałym przykurczem naczyń (co pośrednio ogranicza
zakres metabolicznej regulacji przepływu) ze względu na niedobór NO oraz zwiększoną produkcję endoteliny-1 (ET-1) i wywołany przez nią skurcz naczyń i stres oksydacyjny. W prawidłowym naczyniu, siła ścinająca stymuluje śródbłonkową produkcję NO i ET-1 ale NO
wtórnie hamuje uwalnianie ET-1. Dlatego w prawidłowym naczyniu, efektem zwiększonego
przepływu jest uwalnianie NO i rozkurcz naczynia, bez wyraźnych zmian uwalniania ET-1.
W naczyniu z dysfunkcją, uwalnianie ET-1 nie jest hamowane przez NO i wzrost siły ścinającej może skutkować paradoksalnym skurczem ściany naczyniowej a nie rozkurczem. Aminy
katecholowe, acetylocholina, serotonina, ADP, histamina, endotelina-1, działają rozkurczająco lub kurcząco na naczynia w zależności od stanu funkcjonalnego śródbłonka. Działają one
193
równocześnie na śródbłonek, gdzie stymulują produkcję NO, i bezpośrednio na mięśnie gładkie, gdzie pobudzają skurcz. W naczyniu ze zdrowym śródbłonkiem ich działanie wynikające z uwalniania NO przeważa i następuje rozkurcz naczynia. W naczyniach z uszkodzonym
śródbłonkiem powodują skurcz naczynia [1].
IX.4.2. Działanie przeciwzapalne i przeciwmiażdżycowe śródbłonka
Czynnikiem warunkującym rozwój naczyniowych zmian miażdżycowych i powodującym destabilizację już ukształtowanych blaszek miażdżycowych, jest przewlekły proces zapalny toczący się w intimie dużych tętnic. Aktualna hipoteza na temat mechanizmu powstawania miażdżycy zakłada, że proces jest odpowiedzią ściany naczyniowej na uszkodzenie śródbłonka naczyniowego (response-to injury hypothesis). Badania eksperymentalne potwierdziły,
że śródbłonkowy NO zapobiega rozwojowi naczyniowych zmian miażdżycowych i że stanowi
ważny element ochrony antymiażdżycowej naczyń. Na prawdopodobną sekwencję miażdżycorodnych wydarzeń składają się następujące elementy [25]:
1. Uszkodzenie śródbłonka naczyniowego;
2. Zwiększona naczyniowa produkcja ROS i inaktywacja NO, których rezultatem są:
a. Aktywacja fenotypu prozapalnego śródbłonka poprzez aktywację czynnika transkrypcyjnego NF-κB bezpośrednio przez ROS jak i wtórnie do niedoboru NO. Elementem
fenotypu zapalnego jest między innymi zwiększona ekspresja molekuł adhezyjnych na
powierzchni śródbłonka (selektyny E i P, ICAM-1, VCAM-1) oraz chemokin o działaniu
chemotaktycznym w stosunku do komórek zapalnych (np. MCP-1). W zmianach miażdżycowych ekspresja wszystkich wymienionych molekuł adhezyjnych jest zwiększona.
Jednakże dla rozwoju naczyniowych zmian miażdżycowych (przynajmniej w mysim modelu miażdżycy) szczególnie ważna jest zwiększona ekspresja jedynie obu selektyn oraz
VCAM-1;
b. Oksydacyjna modyfikacja LDL i dalsza aktywacja śródbłonka przez ox-LDL;
3. Adhezja i transmigracja monocytów do i przez warstwę śródbłonka a następnie przekształcanie się ich w makrofagi i ich gromadzenie makrofagów w intimie w miejscu uszkodzenia
śródbłonka;
4. Endocytoza ox-LDL przez makrofagi przy udziale receptorów wymiatających (np. SR-A oraz
LOX-1) i powstawanie oraz gromadzenie się komórek piankowatych w intimie;
5. Zmiana fenotypu, proliferacja i migracja komórek mięśni gładkich ściany naczyniowej
i powstawanie torebki łącznotkankowej blaszki miażdżycowej.
Uszkodzenie śródbłonka i aktywacja jego prozapalnego fenotypu mogą być skutkiem
działania zarówno tradycyjnych czynników ryzyka miażdżycy jak i zaburzeń lokalnego przepływu krwi. W obu przypadkach dochodzi do naczyniowej nadprodukcji O2– i spadku biodostępności śródbłonkowego NO. Oba te czynniki skutkują aktywacją NFκB i prozapalnym
fenotypem śródbłonka, gdyż w stanach, w których biodostępność NO jest upośledzona, NFkB nie jest zwrotnie hamowany przez NO (ryc. 9.2.).
Na rozwój zmian miażdżycowych szczególnie narażone są te miejsca w układzie tętniczym, w których przepływ krwi, zamiast laminarnego, jest albo bardzo mały albo burzliwy
albo pulsacyjny, co oznacza brak lub bardzo małą siłę ścinającą i mały poziom aktywacji
eNOS. W tym kontekście wykazano, że w przeciwieństwie do przepływu laminarnego, prze194
pływ burzliwy i/lub pulsacyjny zwiększają śródbłonkową ekspresję mRNA różnych prozapalnych czynników, w tym VCAM-1, ICAM-1 i E-selektyny i że efektowi temu zapobiega
antyoksydant N-acetylcysteina [5].
Innym ważnym elementem powstawania i dojrzewania blaszki miażdżycowej jest proliferacja i migracja komórek mięśni gładkich ściany naczyniowej do ogniska zapalenia. NO
hamuje proliferację oraz migrację komórek mięśni gładkich zarówno in vitro jak i in vivo i jest
to działanie zależne od aktywacji cyklazy guanylowej. Wykazano, że suplementacja L-argininy zmniejsza restenozę po angioplastyce [22] a donory NO ograniczają proliferację neointimy
w modelu doświadczalnym miażdżycy u królika [3], świni [24] i szczura [9].
IX.4.3. Działanie przeciwkrzepliwe i prokoagulacyjne śródbłonka
Groźnym powikłaniem procesu miażdżycowego są ostre zespoły wieńcowe, których mechanizm obejmuje; (i) uszkodzenie śródbłonka i/lub otoczki łącznotkankowej pokrywających
blaszkę miażdżycową oraz (ii) powstawanie wewnątrznaczyniowego zakrzepu w miejscu
uszkodzenia blaszki miażdżycowej. Czynniki zwiększające krzepliwość krwi są czynnikami
ryzyka wystąpienia ostrych zespołów wieńcowych, w tym zawału serca.
Prawidłowy śródbłonek na drodze przynajmniej czterech mechanizmów zapobiega
powstawaniu zakrzepów w tętnicach: (i) separuje krew od prokrzepliwej macierzy pozakomórkowej; (ii) NO i prostacyklina produkowane przez prawidłowy śródbłonek hamują adhezję, agregację i aktywację płytek krwi; (iii) prawidłowy śródbłonek, którego glikokaliks jest
nieuszkodzony, przeciwdziała produkcji prokoagulacyjnej trombiny oraz (iv) prawidłowy
śródbłonek produkuje duże ilości tkankowego (t-PA) i urokinazowego (u-PA) aktywatora
plazminogenu i niewielkie ilości inhibitora t-PA (PAI-I), dzięki czemu przewagę zdobywa
aktywność fibrynolityczna śródbłonka [27].
IX.5. Kliniczne metody pomiaru czynności śródbłonka
Naczyniorozszerzające, przeciwzapalne, przeciwkrzepliwe i antymiażdżycowe działania
śródbłonka zależą głównie od śródbłonkowej produkcji NO. Nie dysponujemy żadną użyteczna metodą bezpośredniego pomiaru NO. Dlatego w ocenie śródbłonka i śródbłonkowej
produkcji NO posługujemy się jedynie pośrednimi czynnościowymi lub biochemicznymi
wskaźnikami.
Najczęściej stosowane czynnościowe metody oceny śródbłonka opierają się na założeniu, że pomiar naczyniorozszerzającego działania śródbłonka jest dobrą miarą śródbłonkowej biodostępności NO i pośrednio stanu czynnościowego śródbłonka jako całości. W eksperymentach zwierzęcych preferowane jest badanie wielkości rozkurczu badanego naczynia lub
wzrostu przepływu w badanym obszarze naczyniowym pod wpływem substancji, o których
wiadomo, że stymulują śródbłonkową produkcję NO (ryc. 9.4.).
Najczęściej stosowaną w klinice metodą badania czynności śródbłonka jest ultrasonograficzna ocena rozszerzania się dużej tętnicy (zwykle ramieniowej, co wymaga użycia głowicy USG o dużej częstotliwości) pod wpływem zwiększonego przepływu krwi (i wobec tego
zwiększonej siły ścinającej) w tej tętnicy (flow mediated dilatation, FMD) a następnie po podaniu nitrogliceryny lub nitroprusydku sodu [6] (ryc. 9.4.).
195
Ryc. 9.4. Czynnościowe metody oceny śródbłonka. (I) Pomiar rozkurczu naczynia pod wpływem agonisty (np. acetylocholiny, ACh), o którym wiadomo, że zwiększa uwalnianie NO przez śródbłonek.
Aktywacja receptorów ACh na śródbłonku skutkuje uwolnieniem śródbłonkowego NO i rozkurczem
naczynia natomiast równoczesna aktywacja receptorów ACh na mięśniach gładkich naczyń powoduje
ich skurcz. Ostateczny efekt naczyniowy ACh jest wynikiem sumowania się tych dwóch przeciwstawnych efektów. W naczyniu z prawidłowym śródbłonkiem (B), ma miejsce rozkurcz naczynia, bo efekt
śródbłonkowy przeważa. W naczyniu z dysfunkcją śródbłonka rozkurcz jest w różnym stopniu osłabiony lub nawet naczynie się przykurcza (C), bo przeważa efekt działania ACh na mięśniówkę gładką.
(II) Pomiar rozkurczu naczynia prowokowanego zwiększonym przepływem i siłą ścinającą (np. pod
wpływem reaktywnej hiperemii w dorzeczu badanej tętnicy). W naczyniu z prawidłowym śródbłonkiem, wzrostowi przepływu towarzyszy wzrost średnicy naczynia. W naczyniu z dysfunkcja śródbłonkową, taki sam wzrost przepływu, skutkuje mniejszym rozkurczem lub nawet zwężeniem naczynia.
W metodzie pomiaru FMD, przy pomocy mankietu do pomiaru ciśnienia, zaciska się na
klika minut tętnicę na kończynie, co powoduje jej niedokrwienie i związany z tym rozkurcz
naczyń mikrokrążenia. Następnie zwalnia się ucisk, co skutkuje zwiększonym przepływem
krwi i zwiększoną siłą ścinającą w całym dorzeczu zaciśniętej poprzednio tętnicy. W warunkach prawidłowych towarzyszy temu rozkurcz badanej tętnicy (pomiar powyżej miejsca ucisku). Wykazano, że reakcję tę blokują inhibitory syntazy NO, co dowodzi, że jest ona spowodowana NO uwalnianym w wyniku zwiększonego przepływu/siły ścinającej. W naczyniach
z małą produkcją lub dostępnością NO, zwiększony przepływ, prowokuje mniejszy rozkurcz
tętnicy, brak zmian jej średnicy, lub nawet jej skurcz. Przyczyną tych zaburzeń rozkurczu
może być mała dostępność i/lub produkcja NO lub zmniejszona wrażliwości naczynia na
rozkurczające działanie NO. Dlatego elementem każdego badania jest ocena reakcji badanej
tętnicy na doustne podanie nitrogliceryny lub nitroprusydku sodu (egzogenne donory NO).
Metoda FMD jest preferowaną metodą oceny śródbłonka ze względu na jej nieinwazyjności
i niewielkie wymagania sprzętowe. Wymaga jednak udziału dobrze wyszkolonego i doświadczonego operatora, co jest jej poważnym ograniczeniem.
196
Inna metoda oceny rozkurczu naczyń zależnego od śródbłonka polega na dotętniczym
podawaniu agonisty, o którym wiadomo, że zwiększa śródbłonkową produkcję NO. Używane
w tym celu substancje, takie jak acetylocholina, serotonina czy substancja P, obok działania
na śródbłonek (naczynio-rozkurczającego) mają bezpośrednie działanie kurczące na komórki
mięśni gładkich naczyń. Dlatego ostateczny efekt ich działania jest wypadkową tych dwóch
efektów. W naczyniu ze zdrowym śródbłonkiem wymienione substancje powodują rozkurcz,
a w naczyniu z upośledzoną biodostępnością i/lub produkcją NO brak jest efektu rozkurczowego lub dochodzi nawet do skurczu naczynia. Wyniki badań porównawczych sugerują, że
metody z agonistą większa wartość rokowniczą niż metoda FMD. Ograniczeniem metody jest
konieczność dożylnego podawania agonistów.
Do czynnościowych metod oceny funkcji śródbłonka należy także pomiar szybkości fali
tętna. Im większa jest sztywność tętnicy tym szybciej rozchodzi się w niej fala tętna. Wiadomo, że sztywność tętnic częściowo zależy od funkcji śródbłonka i lokalnej produkcji NO.
Badania porównawcze pokazują, że wyniki pomiaru fali tętna korelują z wynikami badania
FMD. Zaletą pomiaru fali tętna jest nieinwazyjność metody. Jednakże pomiar ten informuje
o śródbłonku jedynie w bardzo pośredni sposób, co jest wadą metody i co jest prawdopodobną przyczyną jej niskiej wartości rokowniczej.
Istnieją próby oceny czynności śródbłonka w oparciu o markery biochemiczne mierzone
we krwi. Metody biochemiczne są jednak znacznie gorzej wystandaryzowane niż czynnościowe. Spośród markerów dysfunkcji śródbłonka najczęściej mierzone są poziomy rozpuszczalnych cząstek adhezyjnych we krwi. W miarę rozwoju dysfunkcji, rośnie na nim ekspresja
cząstek adhezyjnych (P i E selektyny oraz VCAM-1 i ICAM-1), które następnie są „odcinane”
przez enzymy proteolityczne i krążą we krwi w postaci rozpuszczalnej. Innym wskaźnikiem
czynności śródbłonka może być pomiar stosunku stężeń we krwi: tkankowy aktywator plazminogenu (tPA)/inhibitor tPA (PAI-1).
Mierzone są także we krwi lub moczu poziomy produktów utleniania NO, jakimi są azotany i azotyny. Pomiar ten, w istocie, informuje o wielkości produkcji NO, a nie o jego biologicznej dostępności. Obok śródbłonka, istnieją inne potencjalne źródła azotynów/azotanów
w płynach biologicznych (np. pokarmowe, leki), co utrudnia interpretację pomiarów.
IX.6. Choroby i stany kliniczne z dysfunkcją śródbłonkową
Dysfunkcja śródbłonkowa jest najczęściej definiowana jako upośledzenie rozkurczu naczyń zależnego od śródbłonka (w domyśle od śródbłonkowego NO) przy zachowanej ich
zdolności do rozkurczu pod wpływem egzogennego źródła NO, jakim jest nitrogliceryna lub
nitroprusydek. Tak definiowana dysfunkcja śródbłonkowa [7]:
1. W modelach eksperymentalnych – była czynnikiem przyspieszającym bądź wyzwalającym
rozwój miażdżycy, nadciśnienia tętniczego i niewydolności serca;
2. Mierzona na tętnicy ramieniowej u ludzi – jest reprezentatywna dla stanu funkcjonalnego
całego układu tętniczego, w tym tętnic wieńcowych, co dowodzi, że zaburzenia funkcji
śródbłonka są procesem ogólnoustrojowym;
3. Była opisywana w licznych stanach i schorzeniach (tab. 9.3.), w tym powszechnie towarzyszy czynnikom ryzyka choroby sercowo-naczyniowej (hipercholesterolemią, cukrzy197
ca, nadciśnienie, otyłość) jak i samej chorobie wieńcowej, co sugeruje, że czynniki ryzyka
przyczyniają się do postępu choroby sercowo-naczyniowej właśnie poprzez upośledzanie
czynności śródbłonka;
Tabela 9.3. Stany i schorzenia przebiegające z dysfunkcją śródbłonka [7]
Stan
Starzenie
Otyłość
Upośledzona tolerancja glukozy
Insulinooporność
Hipercholesterolemia
Hipertriglicerydemia
Hiperhomocysteinemia
Poposiłkowa lipemia
Palenie papierosów
Stan po menopauzie
Zapalenie
Niska urodzeniowa masa ciała
Uraz
Stres psychiczny
Zespół bezdechu sennego
Dializoterapia
Stosowanie glikokortykoidów,
Tolerancja na azotany
Stent uwalniający lek przeciwproliferacyjny
Schorzenia
Choroba wieńcowa
Niewydolność serca
Nadciśnienie tętnicze
Niewydolność nerek
Cukrzyca typu 1 i 2
Stan przedrzucawkowy
Zapalenie naczyń
Reumatoidalne zapalenie stawów
Choroby przyzębia
Posocznica
4. W kilkunastu dotychczasowych prospektywnych badaniach klinicznych dotyczących bardzo różnych grup pacjentów okazała się czynnikiem ryzyka występowania zdarzeń sercowo-naczyniowych i udaru mózgu (im dysfunkcja większa tym większe ryzyko zdarzeń)
[28]. Znaczenie prognostyczne dysfunkcji jest szczególnie duże w grupie osób bez klasycznych czynników ryzyka, co podkreśla użyteczność oceny śródbłonka w procesie stratyfikacji ryzyka [12];
5. Jeżeli uległa poprawie w wyniku leczenia przeciwnadciśnieniowego u kobiet z menopauzą
to było to związane z mniejszym ryzykiem występowania incydentów sercowo-naczyniowych (im większa poprawa funkcji śródbłonka tym lepsza prognoza) [26];
6. U pacjentów z kardiologicznym zespołem X, dysfunkcji śródbłonkowej towarzyszą zaburzenia perfuzji miokardium w scyntygrafii wysiłkowej a poprawie czynności śródbłonka
pod wpływem statyn towarzyszy poprawa perfuzji [7];
7. Jest czynnikiem ryzyka szybkości postępu niewydolności nerek [23], co sugeruje, że niedobór NO ma niekorzystny wpływ na biologie kłębków nerkowych.
Podsumowując, badania eksperymentalne dowodzą, że dysfunkcja śródbłonkowa jest
czynnikiem inicjującym rozwój pierwotnych zmian miażdżycowych w naczyniach. Badania
kliniczne sugerują, że bierze ona udział także w destabilizacji już dojrzałych blaszek miażdżycowych, która to destabilizacja leży u podstawy mechanizmu powstawania ostrych zespołów wieńcowych [7]. O tym ostatnim świadczy fakt, że pomiar funkcji śródbłonka okazał się
198
niezłym pomocniczym testem pozwalającym na wyselekcjonowanie osób ze zwiększonym
ryzykiem wystąpienia ostrych zespołów wieńcowych czy udaru mózgu. Jednakże powyższe
wnioskowania kliniczne są oparte wyłącznie o korelacje statystyczne, które, jako takie, nie są
dowodem na istnienie związku przyczynowo skutkowego między złym rokowaniem i uszkodzeniem śródbłonka.
IX.7. Mechanizm klinicznych postaci dysfunkcji śródbłonkowej
Prawdopodobnie najważniejszym powodem dysfunkcji jest inaktywacja NO przez O2–,
powstający w nadmiarze w naczyniach, co upośledza biologiczną dostępność NO (ryc. 9.1.)
a równocześnie jest źródłem toksycznego nadtlenoazotynu. Innym powodem mogą być różne
zaburzenia wytwarzania NO przez eNOS.
IX.7.1. Nadmierna inaktywacja NO
Normalne naczynia produkują niewielkie ilości O2– w sposób konstytutywny. Produkcja ta
wzrasta w obecności czynników ryzyka sercowo-naczyniowego [4]. Wśród poznanych źródeł
odpowiedzialnych za naczyniową produkcję O2– i stres oksydacyjny najważniejsze to: naczyniowa oksydaza NADPH, oksydaza ksantynowa i eNOS w postaci rozprzęgniętej (rozdz. VIII).
W tym kontekście, na szczególną uwagę zasługuje oksydaza NADPH. Po pierwsze jej aktywność jest regulowana przez czynniki mające związek z powstawaniem i rozwojem miażdżycy jak: hipercholesterolemia, angiotensyna II, czy prozapalne cytokiny. Po drugie, przynajmniej
w takich stanach jak zwiększony przepływ pulsacyjny, reperfuzyjne uszkodzenie śródbłonka,
naczyniowy stres oksydacyjny pod wpływem endoteliny [18] czy kardiomiopatia spowodowana kokainą [16], NADPH kontroluje aktywność oksydazy ksantynowej (ryc. 8.3.).
Angiotensyna II aktywuje NADPH oksydazę i naczyniową produkcję O2– poprzez aktywację receptora angiotensynowego AT1. Czynnikami zwiększającymi naczyniową ekspresję
receptorów AT1 są między innymi takie czynniki jak zwiększone poziomy LDL, interleukina IL-6, cukrzyca czy niedobór estrogenów. Wszystkie te czynniki zwiększają naczyniową
produkcję O2–, wywołują dysfunkcję śródbłonkową i mają działanie miażdżycorodne w różnych modelach eksperymentalnych. Stąd hipoteza, że zwiększona aktywność receptorów AT1
i związana z tym naczyniowa nadprodukcja O2– (prawdopodobnie przez naczyniową NADPH
oksydazę) są główną przyczyną stresu oksydacyjnego i dysfunkcji śródbłonkowej, przynajmniej u osób z chorobą wieńcową [4]. W tym kontekście wykazano w różnych modelach
zwierzęcych, że blokada receptorów AT1, a także zahamowanie NADPH oksydazy przez statyny skutkuje zahamowaniem naczyniowej produkcji O2– i poprawą funkcji śródbłonka [30].
Wykazano ponadto, że w naczyniach izolowanych od ludzi, w tym w tętnicach wieńcowych, NADPH oksydaza jest głównym źródłem naczyniowej produkcji O2–, i że aktywność
tego enzymu, naczyniowa produkcja O2– i dysfunkcja śródbłonkowa korelują z nasileniem
czynników ryzyka choroby sercowo-naczyniowej [11].
Rolę stresu oksydacyjnego i dysfunkcji śródbłonkowej w rozwoju choroby sercowo-naczyniowej ilustruje klasyczne badanie Heitzer’a i wsp. [14]. Uczestniczyli w nim pacjenci z potwierdzoną chorobą wieńcową. U wszystkich oceniano funkcję śródbłonka a także wpływ dożylnego
podania kwasu askorbinowego (użytego jako wymiatacza wolnych rodników) na dysfunkcję
199
śródbłonkową. Wieloletnia prospektywna obserwacja wykazała, że występowanie incydentów
sercowo-naczyniowych było znamiennie większe u osób z bardziej nasiloną dysfunkcją śródbłonkową, ale tylko tych, u których kwas askorbinowy skutkował natychmiastową poprawą
czynności śródbłonka (dowód na wolnorodnikowy mechanizm dysfunkcji). Wniosek z tego
badania klinicznego jest taki, że dysfunkcja śródbłonkowa, zwłaszcza ta spowodowana stresem
oksydacyjnym jest czynnikiem ryzyka incydentów sercowo-naczyniowych u osób z chorobą
wieńcową. Ta ostatnia obserwacja sugeruje ponadto, że stres oksydacyjny może wpływać na
aktywność choroby wieńcowej także w jakiś sposób niezależny od dysfunkcji śródbłonkowej.
IX.7.2. Zmniejszone wytwarzanie NO
Istnieją sprzeczne doniesienia na temat poziomu ekspresji białka eNOS w stanach traktowanych jako czynniki ryzyka sercowo-naczyniowego. W hipercholesterolemii i łagodnych
postaciach choroby wieńcowej zmian ekspresji eNOS nie obserwowano. Natomiast ekspresja
eNOS była zmniejszona w zaawansowanych postaciach choroby wieńcowej i w zaawansowanych zmianach miażdżycowych [8].
W modelach zwierzęcych cukrzycy typu I obserwowano zarówno wzrost (na początku
choroby [15] jak i późniejszy spadek ekspresji eNOS [21]. Pod wpływem m.in. cholesterolu
LDL może dochodzić do wzrostu ekspresji wewnątrzkomórkowego białka kaweoliny-1, które
wiążąc eNOS z błoną komórkową, osłabia jej funkcję.
Niedobór L-argininy; Potencjalną przyczyną zmniejszonej produkcji NO może być niedobór substratu do produkcji NO – L-argininy. Nie obserwowano znacznych zmian poziomów
L-argininy we krwi w stanach, którym towarzyszy dysfunkcja śródbłonkowa. Możliwe jest natomiast istnienie defektów transportu dokomórkowego L-argininy, gdzie jest czynnie transportowa przez kationowy transporter aminokwasów 1 (CAT 1). W badaniach in vitro, jak i in vivo
pokazano, że suplementacja L-argininy skutkująca wzrostem jej stężenia w osoczu prowadzi do
wzrostu wytwarzania NO i poprawy funkcji śródbłonka. Pokazano także, że cholesterol LDL,
a zwłaszcza ox-LDL, hamują transport L-argininy, co prowadzi do czynnościowego niedoboru
L-argininy. W różnych stanach patologicznych, w tym w cukrzycy typu 2, może rosnąć aktywność komórkowej arginazy-1, enzymu rozkładającego L-argininę, co może prowadzić do wewnątrzkomórkowego niedoboru aminokwasu. W jednym badaniu u ludzi pokazano, że krótkotrwała, jak i długotrwała (do 6 miesięcy) suplementacja L-argininy w diecie ma korzystny
wpływ na dysfunkcję śródbłonka [20]. Jednak w innym badaniu, podobna 6-miesięczna suplementacja L-argininy towarzysząca leczeniu choroby naczyń obwodowych, spowodowała pogorszenie funkcji śródbłonka i skrócenie dystansu chromania [31]. Interpretację efektów L-argininy komplikuje fakt, że jest ona dodatkowo stymulatorem uwalniania insuliny. Konkludując, rola
l-argininy w indukcji i leczeniu dysfunkcji śródbłonka jest niepewna.
Niedobór tetrahydrobiopteryny (BH4) [17]; BH4 jest kofaktorem eNOS i jest substancją
łatwo degradowaną przez ROS, a zwłaszcza przez nadtlenoazotyn. Niedobór BH4 powoduje
rozprzęganie eNOS i enzym zwiększa wtedy produkcję O2– kosztem produkcji NO (ryc. 9.3.).
Substratem do komórkowej produkcji BH4 jest GTP.
Krótkotrwała suplementacja BH4 u palaczy i osób z hipercholesterolemią, chorobą wieńcową, niewydolnością serca, cukrzycą typu 2 czy nadciśnieniem skutkowała poprawą naczynio-rozkurczającej czynności śródbłonka. W niedawnym badaniu wykazano, że 4-tygodnio200
wa suplementacja BH4 skutkowała poprawą czynności śródbłonka u osób z hipercholesterolemią, natomiast pozostaje bez wpływu na czynność śródbłonka u osób z prawidłowym stężeniem cholesterolu. Dodatkowo, w hodowli komórek śródbłonka, BH4 zmniejszała stężenie
markerów stresu oksydacyjnego we krwi i zapobiegała rozprzęganiu eNOS pod wpływem
LDL cholesterolu. Interpretację badań z BH4 komplikuje jednak fakt, że substancja ta jest
dodatkowo silnym antyoksydantem. Genetycznie uwarunkowane zaburzenia szlaku BH4 są
czynnikiem ryzyka rozwoju chorób układu sercowo-naczyniowego.
Zahamowanie eNOS przez niesymetryczną dimetyloargininę (ADMA) [2]; ADMA jest
pochodną L-argininy i endogennym inhibitorem eNOS. Powstaje w reakcji katalizowanej przez
metylotransferazy z udziałem metylowanych białek i jest usuwana przez nerki z moczem. Stres
oksydacyjny jest czynnikiem aktywującym enzym produkujący i hamującym enzym rozkładający ADMA. ADMA działając na eNOS rozprzęga ten enzym, czemu towarzyszy spadek
produkcji NO i wzrost produkcji O2–. W sumie więc produkcja ADMA jest wtórna do stresu
oksydacyjnego i dodatkowo, na drodze sprzężenia zwrotnego, stres ten wzmaga. Podwyższone
stężenia ADMA we krwi stwierdzano u osób z hipercholesterolemią, niewydolnością nerek,
nadciśnieniem tętniczym, cukrzycą i hiperhomocysteinemią i stężenia te korelowały z nasileniem dysfunkcji śródbłonka, co sugeruje udział ADMA w rozwoju dysfunkcji śródbłonkowej.
Podwyższone stężenie ADMA w osoczu jest niezależnym czynnikiem ryzyka progresji miażdżycy, umieralności z powodów sercowo-naczyniowych i umieralności całkowitej. Inhibitory
konwertazy angiotensyny, metformina i aspiryna obniżają stężenie ADMA we krwi.
IX.8. Interwencje lecznicze o działaniu śródbłonkowym
Najskuteczniejszą interwencją poprawiającą funkcję śródbłonka jest wysiłek fizyczny.
Regularny wysiłek fizyczny, poprzez zwiększanie przepływu krwi i siły ścinającej, zwiększa
podstawową i stymulowaną produkcję NO i czynność śródbłonka u zwierząt eksperymentalnych i u ludzi zdrowych oraz z różnymi chorobami układu sercowo-naczyniowego [10] na
drodze czterech mechanizmów:
1. zwiększa aktywność i/lub ekspresję eNOS;
2. zwiększa naczyniową ekspresję enzymów antyrodnikowych (SOD, katalaza, peroksydaza
glutationu);
3. zmniejsza ekspresję naczyniowych enzymów produkujących O2– (oksydaza NADPH, oksydaza ksantynowa) oraz
4. zwiększa liczbę śródbłonkowych komórek progenitorowych (EPC) we krwi.
Podobny efekt obserwowano u świń z podwiązką imitującą blaszkę miażdżycową, w przewlekle niedokrwionym obszarze i krążeniu obocznym oraz u pacjentów z przewlekłą chorobą
wieńcową [13].
Interwencje hipolipemizujące; Wszystkie badane interwencje hipolipemizujące (cholestyramina, LDL-afereza, ezetymib) zmniejszały dysfunkcję śródbłonkowa, co sugeruje, że
sama redukcja stężenia cholesterolu całkowitego i/lub cholesterolu LDL we krwi prowadzi
do poprawy funkcji śródbłonka. Szczególnie silny korzystny wpływ na funkcję śródbłonka
mają statyny, co, przynajmniej częściowo, ma związek z ich działaniem plejotropowym, a nie
koniecznie hipolipemizującym.
201
Statyny poprawiają funkcję śródbłonka u zdrowych osób z hipercholesterolemią, stabilną chorobą wieńcową, po przebytym ostrym zespole wieńcowym [7]. U pacjentów z hipercholesterolemią i zaburzeniami perfuzji w scyntygrafii wysiłkowej serca pokazano, że po 12
tygodniach podawania fluwastatyny doszło do poprawy funkcji śródbłonka, czemu towarzyszył istotny wzrost perfuzji segmentów niedokrwionych w wyjściowym badaniu [7]. Dwie
obserwacje wskazują, że istotnym elementem we wpływie statyn są tzw. działania plejotropowe, czyli nie związane z wpływem na stężenie lipidów w surowicy krwi: zmiany funkcji
śródbłonka nie korelują ze spadkiem stężenia całkowitego cholesterolu i cholesterolu LDL,
a korzystny wpływ atorwastatyny na funkcję śródbłonka pojawiał się już po 1 dniu stosowania leku, znacznie wyprzedzając zmiany w stężeniu lipidów w surowicy krwi. Postulowane
mechanizmy działania statyn to:
1. Związane z wpływem na stężenie lipidów w surowicy; spadek stężenia cholesterolu LDL
cholesterolu może zmniejszać hamowanie komórkowego transportera L-argininy (CAT-1)
i poprawiać jej dokomórkowy transport;
2. Niezwiązane z wpływem na stężenie lipidów w surowicy (działania plejotropowe):
a. wzrost ekspresji eNOS (poprzez zwiększenie stabilności mRNA dla eNOS);
b. wzrost aktywności eNOS (poprzez fosforylację eNOS przez kinazę Akt);
c. zmniejszenie ekspresji oksydazy NADPH, głównego źródła naczyniowych ROS;
d. zmniejszenie ekspresji kaweoliny-1, kotwiczącej eNOS w błonie komórkowej i hamującej
jej aktywność.
Statyna + losartan silniej poprawiały funkcję śródbłonka, niż każdy z leków z osobna [19].
Fibraty (aktywatory receptora aktywatora proliferatora peroksysomów alfa, PPAR-α),
poprawiają funkcję śródbłonka, zmniejszają nasilenie stresu oksydacyjnego i równocześnie
zwiększają stężenie adiponektyny i insulinowrażliwość tkanek.
Interwencje antyangiotensynowe; W klinice najczęściej stosowane są: (i) inhibitory enzymu konwertującego angiotensynę (IKA) oraz (ii) antagoniści receptora angiotensynowego
AT1 (ARA). Chociaż obie grupy leków blokują wpływ angiotensyny II na tkanki docelowe,
istnieją między nimi istotne różnice. IKA blokują przekształcanie angiotensyny I w angiotensynę II i dodatkowo są inhibitorami rozkładu bradykininy, która aktywując swoje śródbłonkowe receptory, zwiększa śródbłonkową produkcję NO. Natomiast ARA, obok wybiórczego
blokowania receptora AT1, zwiększają stężenie angiotensyny II i jej oddziaływanie z receptorem AT2. Aktywacja śródbłonkowych receptorów AT2 skutkuje zwiększoną śródbłonkową
produkcją NO. Wobec tego, zarówno IKA jak i ARA mają dodatkowe efekty niezwiązane z ich
anty-angiotensynowym działaniem. IKA i ARA z podobną skutecznością poprawiają funkcję
śródbłonka u pacjentów z chorobą wieńcową, niewydolnością serca, cukrzycą, nadciśnieniem
tętniczym. Postulowane mechanizmy tego działania to:
1. mniejszy stres oksydacyjny: aktywność naczyniowej oksydazy NADPH znajduje się pod
kontrolą receptora angiotensynowego AT1 ;
2. poprawa wrażliwości tkanek na insulinę;
3. mniejsze nasilenie stanu zapalnego.
Witaminy antyoksydacyjne ani hormonalna terapia zastępcza nie poprawiały czynności
śródbłonka w różnych badaniach klinicznych.
202
Piśmiennictwo
1. Beręsewicz A, Mączewski M. Regulacja przepływu wieńcowego w warunkach prawidłowych
i w nadciśnieniu tętniczym. Nadciśnienie tętnicze. Kraków: Medycyna Praktyczna 2007: 355-362.
2. Böger RH, Bode-Böger SM et al. Asymmetric dimethylarginine (ADMA): A novel risk factor for
endothelial dysfunction : Its role in hypercholesterolemia. Circulation 1998; 98:1842-1847.
3. Bult H, De Meyer GR, and Herman AG. Influence of chronic treatment with a nitric oxide donor on
fatty streak development and reactivity of the rabbit aorta. Br J Pharmacol 1995; 114: 1371-82.
4. Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress.
Circ Res 2000; 87:840-4.
5. Chappell DC, Varner SE, Nerem RM, Medford RM, Alexander RW. Oscillatory shear stress stimulates adhesion molecule expression in cultured human endothelium. Circ Res 1998; 82:532-539.
6. Corretti MC, Anderson TJ, Benjamin EJ et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: A report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol 2002; 39:257-65.
7. Davignon J, Ganz P. Role of Endothelial dysfunction in atherosclerosis. Circulation 2004; 109 (Suppl
III):III-27-III-32.
8. Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide
synthase. Am J Physiol 2003; 284:R1-12.
9. Guo JP, Milhoan KA, Tuan RS, and Lefer AM. Beneficial effect of SPM-5185, a cysteine-containing
nitric oxide donor, in rat carotid artery intimal injury. Circ. Res. 1994; 75:77-84.
10. Gielen S, Schuler G, Adams V. Cardiovascular Effects of exercise training: molecular mechanisms.
Circulation 2010; 122:1221-38.
11. Guzik TJ, West NE, Black E et al. Vascular superoxide production by NAD(P)H oxidase: association
with endothelial dysfunction and clinical risk factors. Circ Res 2000; 86:E85-E90.
12. Halcox JPJ, Schenke WH, Zalos G et al. Prognostic value of coronary vascular endothelial dysfunction. Circulation 2002; 106:653-8.
13. Hambrecht R, Adams V, Erbs S et al. Regular physical activity improves endothelial function in
patients with coronary artery disease by increasing phosphorylation of endothelial nitric oxide synthase. Circulation 2003; 107:3152-8.
14. Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T. Endothelial dysfunction, oxidative stress, and risk
of cardiovascular events in patients with coronary artery disease. Circulation 2001; 104:2673-2678.
15. Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res 2001; 88:E14-E22.
16. Isabelle M, Vergeade A, Moritz F et al. NADPH oxidase inhibition prevents cocaine-induced up-regulation of xanthine oxidoreductase and cardiac dysfunction. J Mol Cell Cardiol 2007; 42:326-332.
17. Katusic ZS, d’Uscio LV, Nath KA. Vascular protection by tetrahydrobiopterin: progress and therapeutic prospects. Trends in Pharmacological Sciences 2009; 30:48-54.
18. Klemenska E, Konior A, Beresewicz A. ET-1-induced O2- generation and endothelial dysfunction
in guinea-pig heart; Role of PKC, mirochondria and NADPH oxidase and xanthine oxidase. J.Mol.
Cell Cardiol. 2008; 44:727.
19. Koh KK, Quon MJ, Han SH et al. Additive beneficial effects of losartan combined with simvastatin in
the treatment of hypercholesterolemic, hypertensive patients. Circulation 2004; 110:3687-92.
20. Lerman A, Burnett JC, Jr., Higano ST, McKinley LJ, Holmes DR, Jr. Long-term L-arginine supplementation improves small-vessel coronary endothelial function in humans. Circulation 1998;
97:2123-8.
203
21. Nagareddy PR, Xia Z, Mcneill JH, Macleod KM. Increased expression of iNOS is associated with
endothelial dysfunction and impaired pressor responsiveness in streptozotocin-induced diabetes.
Am J Physiol 2005; 289:H2144-H2152.
22. Niebauer J, Schwarzacher SP, Hayase M et al. AC. Local L-arginine delivery after balloon angioplasty
reduces monocyte binding and induces apoptosis. Circulation 1999; 100:1830-1835.
23. Perticone F, Maio R, Perticone M et al. Endothelial dysfunction andsubsequent decline in glomerular filtration rate in hypertensive patients. Circulation 2010; 122:379-84.
24. Provost P, Tremblay J, and Merhi Y. The antiadhesive and antithrombotic effects of the nitric oxide
donor SIN-1 are combined with a decreased vasoconstriction in a porcine model of balloon angioplasty. Arterioscler Thromb Vasc Biol 1997; 17:1806-1812.
25. Ross R. Cell biology of atherosclerosis. Annu Rev Physiol 1995; 57:791-804.
26. Rossi R, Nuzzo A, Origliani G, Modena MG. Prognostic role of flow-mediated dilation and cardiac
risk factors in post-menopausal women. J Am Coll Cardiol 2008; 51:997-1002.
27. Simionescu M. Implications of early structural-functional changes in the endothelium for vascular
disease. Arterioscler Thromb Vasc Biol 2007; 27:266-274.
28. Targonski PV, Bonetti PO, Pumper GM, Higano ST, Holmes DR, Jr., Lerman A. Coronary Endothelial Dysfunction Is Associated With an Increased Risk of Cerebrovascular Events. Circulation 2003;
107:2805-9.
29. Uematsu M, Ohara Y, Navas JP et al. Regulation of endothelial cell nitric oxide synthase mRNA
expression by shear stress. Am J Physiol Cell Physiol 1995; 269:C1371-C1378.
30. Wassmann S, Laufs U, Baumer AT et al. HMG-CoA reductase inhibitors improve endothelial dysfunction in normocholesterolemic hypertension via reduced production of reactive oxygen species.
Hypertension 2001; 37:1450-1457.
31. Wilson AM, Harada R, Nair N, Balasubramanian N, Cooke JP. L-Arginine supplementation in peripheral arterial disease: No benefit and possible harm. Circulation 2007; 116:188-95.
204
X. Zespół kardiometaboliczny
Monika DUDA
X.1. Definicja i kryteria diagnostyczne
Otyłość, dyslipidemie, upośledzenie gospodarki węglowodanowej i nadciśnienie tętnicze
należą do czynników zwiększających ryzyko rozwoju chorób sercowo-naczyniowych. Zabu-
Ryc. 10.1. Zespół kardiometaboliczny (ZKM) i względne ryzyko wystąpienia towarzyszących mu zaburzeń sercowo-naczyniowych. Meta-analizą objęto 87 badań (951 083 pacjentów), których wyniki
opublikowano między 01.2002–06.2009. W badaniach analizowano częstość występowania chorób sercowo-naczyniowych (CVD), lub analizę zawężano do częstości występowania zawału lub udaru. Rozpoznanie ZKM oparto na kryteriach AHA/NHLBI – przy czym w 63 badaniach (497 651 pacjentów) za
hiperglikemię uznano poziom glukozy ≥ 110 mg/dl. N – całkowita liczba osób objętych obserwacją z lub
bez ZKM; n – liczba pacjentów, u których stwierdzono CVD, zawał, udar lub zmarli; RR – względne
ryzyko; Cl – przedział ufności. Na podstawie [1].
205
rzenia te są ze sobą powiązane i często współwystępują, co drastycznie zwiększa ryzyko zachorowań na cukrzycę typu 2 i choroby sercowo-naczyniowe oraz związaną z nimi śmiertelność
(ryc. 10.1.). Stan taki określany jest terminem zespół kardiometaboliczny (ZKM) (synonimy:
zespół metaboliczny, metaboliczny zespół X, śmiertelny kwartet, zespół Raevena).
Trwa dyskusja na temat optymalnych i prostych do zastosowania w praktyce klinicznej
kryteriów rozpoznawania ZKM. Obecnie posługujemy się dwoma zestawieniami takich kryteriów zaproponowanymi przez: (i) The National Cholesterol Education Programme Adult Treatment Panel III z późniejszą modyfikacją American Heart Association/National Heart, Lung,
and Blood Institute (AHA/NHLBI) z 2005 roku oraz (ii) International Diabetes Federation
(IDF) w 2005 roku [2].
Według AHA/NHLBI zespół ZKM pozwala zdiagnozować obecność, co najmniej 3 z 5
wymienionych czynników:
1. otyłość brzuszna mierzona, jako obwód talii ≥102 cm u mężczyzn i ≥ 88 cm u kobiet (pomiar talii nie jest wymagany jeśli BMI ≥30 kg/m2);
2. stężenie triglicerydów ≥150 mg/dl (1.7 mmol/l) lub leczona hipertriglicerydemia;
3. stężenie HDL <40 mg/dl (1.03 mmol/l) u mężczyzn i <50 mg/dl (1.29 mmol/l) u kobiet lub
leczone zaburzenie;
4. glikemia na czczo ≥100 mg/dl (5.6 mmol/l) lub rozpoznana wcześniej cukrzyca typu 2 (jeśli
glikemia na czczo ≥100 mg/dl rekomendowane jest wykonanie testu OGTT);
5. ciśnienie skurczowe ≥130 mmHg i/lub rozkurczowe ≥85 mmHg lub leczone nadciśnienie
tętnicze.
Otyłość brzuszna
(obwód talii: M≥94 cm K≥80 cm)
+
2 czynniki
• wysokie stężenie triglicerydów
(≥150 mg/dl lub leczona hipertrójglicerydemia)
• niskie stężenie HDL
(M<40 mg/dl, K<50 mg/dl lub leczone zaburzenie)
• wysokie stężenie glukozy
(≥100 mg/dl lub rozpoznana wcześniej cukrzyca typu 2)
• nadciśnienie tętnicze
(≥130/85 mmHg lub leczone nadciśnienie tętnicze)
Ryc 10.2. Kryteria rozpoznania zespołu kardiometabolicznego wg IDF z 2005 roku.
Aby rozpoznać ZKM zgodnie z wytycznymi IDF należy stwierdzić otyłość brzuszną oraz co
najmniej 2 z 4 czynników wymienionych na ryc. 10.2. [3]. Dodatkowo jednak zaostrzone zostały
kryteria rozpoznawania otyłości i różnicuje się ją pod względem etnicznym. U Europejczyków
otyłość brzuszną diagnozujemy, gdy obwód talii wynosi ≥ 94 cm u mężczyzn i ≥ 80 cm u kobiet.
206
IDF zwraca również uwagę na inne metody diagnostyczne i parametry biochemiczne,
które lepiej charakteryzują zaburzenia występujące w ZKM i które w przyszłości mogą posłużyć weryfikacji definicji zespołu. Określono je jako „platynowy standard” i należą do niego:
1. ocena rozmieszczenia tkanki tłuszczowej w organizmie przy użyciu CT, MRI lub DEXA
oraz ocena jej tkankowych biomarkerów leptyny i adiponektyny;
2. poziom aterogennych liporotein – nie-HDL cholesterolu (lub białka ApoB) i frakcji małych
LDL;
3. doustny test tolerancji glukozy (OGTT);
4. ocena insulinooporności – poziom insuliny/proinsuliny na czczo, wskaźnik HOMA, test
klamry metabolicznej oraz podwyższony poziom WKT na czczo i w teście Fogtt;
5. ocena dysfunkcji śródbłonka naczyniowego i mikroalbuminuria;
6. markery stanu prozakrzepowego – wysokie stężenie PAI-1 i fibrynogenu;
7. markery odczynu zapalnego – podwyższony poziom CRP, TNFα i IL-6 oraz obniżone stężenie adiponektyny;
8. ocena osi przysadkowo-nadnerczowej.
X.2. Epidemiologia
Badania epidemiologiczne wskazują na znaczne rozpowszechnienie ZKM, szczególnie
w krajach rozwiniętych. Badanie National Health and Nutrition Examination Survey przeprowadzone w latach 1999–2002 w USA pokazuje, że ZKM wg kryteriów AHA/NHLBI występuje
u 33.7% mężczyzn i 35.4% kobiet powyżej 20. roku życia [4]. Przy zastosowaniu zaostrzonych
kryterium IDF oceny otyłości wskaźnik ten wzrasta odpowiednio do 39.9% i 38.1%.
Ryc. 10.3. Częstość występowania zespołu kardiometabolicznego w zależności od płci i wieku w populacji amerykańskiej (A), europejskiej (B) i polskiej (C) [4, 5]. Zastosowanie zaostrzonych kryteriów IDF
prawdopodobnie zwiększyłoby występowanie zespołu kardiometabolicznego w populacji polskiej.
W Europie częstość występowania ZKM jest mniejsza, w zależności od kraju i przyjętych
kryteriów wynosi od 16% do 25% u mężczyzn i od 15% do 21% u kobiet. W Polsce w oparciu
o Wieloośrodowiskowe Ogólnopolskie Badanie Stanu Zdrowia Ludności (WOBASZ) przeprowadzone w latach 2002–2005 z wykorzystaniem kryteriów AHA/NHLBI ZKM dotyczy
23% mężczyzn i 20% kobiet w wieku 20–74 lata [5]. Na częstość występowania ZKM wpły207
wają przynależność do grupy etnicznej, płeć a w szczególności wiek (ryc. 10.3.). Narastająca
w ostatnich latach epidemia otyłości może przyczynić się do wzrostu liczby chorych z ZKM.
Otyłość staje się poważnym problem również wśród dzieci i młodzieży, dlatego ZKM diagnozowany jest w coraz młodszym wieku.
X.3. Rola tkanki tłuszczowej w rozwoju zespołu kardiometabolicznego
Etiologia ZKM pozostaje niewyjaśniona. Teoria ewolucyjna łączy wzrost częstości występowania ZKM z konfliktem, jaki występuje między genotypem Homo sapiens ukształtowanym w trudnej epoce kamienia łupanego a dobrobytem współczesnej epoki (rozdz. I.5) [6]. Zidentyfikowano
polimorfizm ponad 500 000 pojedynczych nukleotydów, które mogą być łączone z zaburzeniami
charakterystycznymi dla ZKM. Najlepiej udokumentowany jest obecnie związek miedzy zaburzeniami tkanki tłuszczowej, jakie towarzyszą współczesnej epidemii otyłości i rozwojem ZKM.
X.3.1. Krótka charakterystyka tkanki tłuszczowej
Komórki tkanki tłuszczowej (adipocyty) nie są jedynie biernym rezerwuarem energii, ale
aktywnie uczestniczą w utrzymaniu stałego poziomu kwasów tłuszczowych i glukozy (pośrednio przez uwalniane substancje) we krwi. W okresie nadmiaru substancji energetycznych
(po posiłku) adipocyty dynamicznie gromadzą zestryfikowane kwasy tłuszczowe w postaci
triglicerydów (lipogeneza), natomiast przy niedoborze substratów energetycznych (na czczo,
w okresie postu) triglicerydy są hydrolizowane (lipoliza) i kwasy tłuszczowe są ponownie
uwalniane do krwi (ryc. 4.5.). Dodatkowo, adipocyty wydzielają szereg czynników o działaniu auto- , para- i endokrynnym, w tym:
1. Adiokiny – hormony o szerokim spektrum działania, z których najlepiej zbadane zostały
leptyna, adiponektyna, rezystyna, apelina i wisfatyna
a. Leptyna jest produkowana głównie przez adipocyty położone podskórnie, proporcjonalnie do ich ilości i wielkości. W osoczu stężenie leptyny koreluje dodatnio z ilością podskórnej tkanki tłuszczowej. Wpływając na ośrodkowy układ nerwowy leptyna zmniejsza
łaknienie oraz zwiększa wydatkowanie energii poprzez stymulację aktywności fizycznej
i termogenezy. Efekty te są wtórne do zahamowania AMPK w hipokampie i aktywacji
układu współczulnego. W tkankach obwodowych leptyna działa poprzez aktywację
AMPK (ryc. 10.4.). W adipocytach zwiększa wychwyt kwasów tłuszczowych i lipogenezę. Poza tkanką tłuszczową nasila lipolizę i β-oksydację kwasów tłuszczowych (m.in.
w mięśniach szkieletowych, mięśniu sercowym) (rozdz. IV). W ZKM rozwija się leptynooporność, która charakteryzuje się upośledzeniem AMPK-zależnego działania hormonu.
Dodatkowo, leptyna jest modulatorem odpowiedzi immunologicznej typu komórkowego – u dzieci z deficytem leptyny znacznie wzrasta częstość infekcji [7]. Pomimo wielu
korzystnych efektów działania leptyny, utrzymujące się jej wysokie stężenie wpływa negatywnie na układ sercowo-naczyniowy.
b. Adiponektyna – głównym jej źródłem, podobnie jak leptyny, jest podskórna tkanka tłuszczowa. W małych ilościach może być produkowana przez kardiomiocyty, komórki śródbłonka i inne. Poziom adiponektyny we krwi koreluje ujemnie z ilością tkanki tłuszczowej,
szczególnie trzewnej. Badania eksperymentalne sugerują, że adipocyty trzewne produkują
208
Ryc. 10.4. Efekty metaboliczne adipokin związane z ich działaniem stymulującym lub hamującym na
kinazy AMP.
cytokiny prozapalne (min. IL-6), które hamują ekspresję genu i produkcję adiponektyny [8].
W osoczu hormon może występować w kilku aktywnych postaciach, jako trimer (LMW,
ang. low-molecular-weight), heksametr, dodekamer i oktadekamer (HMW, ang. high-molecular-weight), które są produktami polimeryzacji oraz jako białko globularne (sferyczne), które
powstaje w wyniku działania enzymów proteolitycznych. Adiponektyna wywiera efekty biologiczne poprzez aktywację receptorów typu 1 (AdipoR1) i typu 2 (AdipoR2) oraz cząsteczki
adhezyjnej – T-kadheryny (T-Cad). Komponenta metaboliczna aktywności adiponektyny
związana jest głównie z aktywacją AMPK, co nasila katabolizm lipidów w mięśniach szkieletowych i wątrobie, i prowadzi do zmniejszenia ilości magazynowanych tam triglicerydów.
Adipokina działa także hipoglikemicznie: zmniejsza produkcję glukozy w wątrobie (glukoneogeneza) oraz zwiększa wychwyt glukozy przez tkanki obwodowe wtórnie do wzrostu
gęstości transporterów glukozy GLUT4 na powierzchni komórek (rozdz. IV). Adiponektyna
jest adipokiną o plejotropowym działaniu kardioprotekcyjnym [9].
c. Rezystyna hamuje aktywność AMPK i prawdopodobnie w tym mechanizmie zwiększa
glukoneogenezę w wątrobie i zmniejsza insulinowrażliwość tkanek. U ludzi, zwiększonym poziomom rezystyny towarzyszy wzrost ryzyka rozwoju cukrzycy typu 2 [10].
d. Wisfatyna – nazwa pochodzi od wisceralnej tkanki tłuszczowej, gdzie jest głównie produkowana. Jej poziom w surowicy dodatnio koreluje z ilością tej tkanki [11]. W przeciwieństwie do innych substancji wydzielanych przez wisceralne adipocyty jest insulinomimetykiem, w tym: zwiększa wychwyt glukozy przez mięśnie szkieletowe i adipocyty,
hamuje uwalnianie glukozy przez hepatocyty oraz zwieksza gromadzenie triglicerydów
w adipocytach. Efekty te są związane z aktywacją receptora insuliny [12].
e. Apelina – prawdopodobnie nie wpływa na metabolizm substratów energetycznych. Są
doniesienia o jej korzystnym działaniu na układ sercowo-naczyniowy.
2. Cytokiny pro-zapalne, w tym: TNFα, IL-1β, IL-6 i białko chemotaktyczne makrofagów
(MCP-1) oraz CRP;
209
3. Białka pro-zakrzepowe, w tym czynnik tkankowy i inhibitor tkankowego aktywatora plazminogenu (PAI-1) hamujący fibrynolizę;
4. Białka układu RAS: renina, angiotensynogen, angiotensyna I, konwertaza angiotensyny
oraz inne proteazy produkujące angiotensynę II.
X.3.2. Otyłość trzewna i ektopowe gromadzenie lipidów
Dodatni bilans energetyczny, wynikający z wysokokalorycznej diety i braku aktywności
fizycznej, prowadzi do wzrostu ilości tkanki tłuszczowej – wzrasta liczba i wielkość adipocytów w organizmie. Nadmierna ilość triglicerydów może być gromadzona w podskórnej i/lub
wisceralnej (trzewnej) tkance tłuszczowej, co prowadzi do rozwoju różnych postaci otyłości.
Podskórna tkanka tłuszczowa uważana jest za bezpieczny magazyn triglicerydów, a otyłość
związana ze wzrostem jej ilości nazywana jest często „zdrową otyłością” (healthy obesity).
U osób z ZKM najprawdopodobniej dochodzi do przeładowania i/lub obniżenia zdolności
magazynowania triglicerydów w podskórnej tkance tłuszczowej. Rolę magazynu lipidów
przejmuje wtedy wisceralna tkanka tłuszczowa, a zjawisku temu sprzyja:
1. Długotrwały dodatni bilans energetyczny;
2. Stres i nadmierna produkcja hormonu adrenokortykotropowego (ACTH) przez przysadkę
mózgową. ACTH pobudza korę nadnerczy do wydzielania kortyzolu, który zwiększa apetyt
na wysokokaloryczne potrawy i sprzyja lipogenezie w wisceralnej tkance tłuszczowej;
3. Leptynooporność, której konsekwencją jest zahamowanie jej lipogenetycznego działania
w podskórnej tkance tłuszczowej;
4. Insulinooporność podskórnej tkanki tłuszczowej, czego konsekwencją jest wzrost jej aktywności lipolitycznej.
Zaburzenia metabolizmu lipidów i glukozy oraz układu sercowo-naczyniowego, charakterystyczne dla zespołu metabolicznego związane są z otyłością wisceralną i są konsekwencją
biologicznej aktywności tej tkanki (ryc. 10.5.):
Ryc. 10.5. Substancje uwalniane do krążenia przez wisceralną tkankę tłuszczową i ich wpływ na inne
narządy.
210
Ze względu na dynamiczny metabolizm i wysoką aktywność lipolityczną niezależną od
insuliny (duża aktywność lipazy lipoproteinowej), trzewna tkanka tłuszczowa jest niestabilnym magazynem triglicerydów i uwalnia nadmierne ilości wolnych kwasów tłuszczowych
do krwioobiegu. Skutkuje to ekotopowym gromadzeniem się lipidów w mięśniach szkieletowych, wątrobie, komórkach β trzustki czy mięśniu sercowym. Coraz powszechniejszy jest
pogląd, że to właśnie lipidy zgromadzone w tkankach obwodowych i ich tzw. lipotoksyczność
są odpowiedzialne za rozwój stanów chorobowych towarzyszących ZKM.
Trzewna tkanka tłuszczowa jest także aktywniejszym niż tkanka podskórna producentem różnych substancji czynnych, w tym: cytokin pro-zapalnych, adiponektyny, czynników
pro-zakrzepowych oraz białek układu RAS, które również mają swój prawdopodobny udział
w rozwoju patologii towarzyszących ZKM.
W badaniach eksperymentalnych wykazano, że przeszczep adipocytów podskórnych do
przestrzeni wewnątrzotrzewnowej znacznie poprawia profil metaboliczny u myszy [13].
X.4. Mechanizm insulinooporności i zaburzeń gospodarki
węglowodanowej
Insulina jest kluczowym anabolicznym hormonem uwalnianym przez komórki β trzustki
w odpowiedzi na poposiłkowy wzrost stężenia glukozy we krwi. Insulina aktywuje szereg
ścieżek sygnalizacyjnych, które zwiększają: (i) komórkowy wychwyt glukozy poprzez wzrost
ekspresji, gęstości w błonach komórkowych i aktywności GLUT1 i GLUT4 (zwłaszcza w mięśniach szkieletowych i adipocytach), (ii) komórkowy wychwyt wolnych kwasów tłuszczowych
poprzez wzrost gęstości w błonach komórkowych transporterów dla kwasów FAT/CD36, oraz
(ii) ilość zmagazynowanych triglicerydów, glikogenu i białek, promując ich syntezę a hamując
odpowiednio glikolizę, lipolizę i katabolizm białek (rozdz. IV).
Otyłości wisceralnej towarzyszy insulinooporność hepatocytów, adipocytów i mięśni szkieletowych. Nie jest natomiast pewne czy dotyczy także mięśnia sercowego i śródbłonka. Zgromadzone w komórkach lipidy, wzrost stężenia pro-zapalnych cytokin i adipokin oraz spadek stężenia adiponektyny upośledzają insulino-zależne ścieżki sygnalizacyjne. W efekcie maleją wychwyt
i zużycie glukozy przez mięśnie szkieletowe i adipocyty i rośnie wątrobowa produkcja glukozy, co
prowadzi do hiperglikemii. Wysokie stężenie glukozy w surowicy prowadzi do kompensacyjnej
hiperinsulinemii. Komórki β trzustki są również wrażliwe na nadmiar lipidów, co prowadzi do
zaburzenia ich funkcji wydzielniczej i w konsekwencji do zahamowania wydzielania insuliny.
X.5. Mechanizm nadciśnienia tętniczego
Mechanizm nadciśnienia tętniczego, jaki towarzyszy ZKM jest zjawiskiem wieloczynnikowym (ryc. 10.6.).
1. Badania eksperymentalne sugerują, że jego ważnym elementem jest hiperleptynemia. Myszy pozbawione genu leptyny są otyłe ale hipotensyjne [14]. Odwrotnie, długotrwała infuzja leptyny podnosi ciśnienie krwi, przy znacznej redukcji ilości spożywanego pokarmu,
co przypisuje się sympatykomimetycznemu działaniu hiperleptynemii [15] i częściowo
hiperinsulinemii;
211
Ryc. 10.6. Mechanizm nadciśnienia w zespole kardiometabolicznym. Wg [16].
2. Innym elementem może być aktywacja układu RAS, wtórna do produkcji jego składowych
przez wisceralną tkankę tłuszczową;
3. Składową mechanizmu jest również nadprodukcja ROS prowadząca do spadku biodostępności NO i dysfunkcji śródbłonka. Źródłem ROS może być naczyniowa (i) oksydaza
NADPH aktywowana przez leptynę, rezystynę i AT II (rozdz. VIII), oraz (ii) mitochondrialny łańcuch oddechowy, o którym wiadomo, że w obecności nadmiaru substratów
energetycznych (kwasów tłuszczowych i glukozy) staje się producentem zwiększonych ilości O2– (rozdz. V.8);
4. Niskie stężenia adiponektyny są związane z upośledzeniem funkcji śródbłonka naczyniowego.
X.6. Związek miażdżycy z zespołem kardiometabolicznym
Mechanizm, w jakim otyłość trzewna i ZKM sprzyjają rozwojowi miażdżycy i jej konsekwencjom sercowo-naczyniowych (ryc. 10.1.) jest poznany jedynie fragmentarycznie. Brane
są pod uwagę następujące niewykluczające się nawzajem mechanizmy:
1. Kwasy tłuszczowe i inne substancje lipidowe są ligandami receptorów wrodzonego układu
odporności i jako takie mogą być bezpośrednimi aktywatorami systemowego i/lub lokalnego odczynu zapalnego, o którym wiadomo, że jest czynnikiem sprzyjającym destabilizacji
blaszek miażdżycowych (rozdz. VII);
2. Otyłości i ZKM towarzyszą zaburzenia profilu lipidowego powszechnie uznawane za miażdżycorodne. Składają się na to wysokie stężenia VLDL1, obecnośc sdLDL i niskie stężenia
HDL. Profil ten jest wynikiem: (i) nadmiernej ilości wolnych kwasów tłuszczowych do produkcji triglicerydów, które trafiają do wątroby bezpośrednio z wisceralnej tkanki tłuszczowej przez żyłę wrotną i (ii) związanej z insulinoopornością zwiększonej syntezy de novo
kwasów tłuszczowych. Duża podaż triglicerydów w wątrobie prowadzi do syntezy VLDL1,
które w osoczu ulegają dalszemu metabolizmowi do sdLDL cechujących się dużą aterogennością, gdyż jako bardzo małe cząsteczki łatwiej penetrują do intimy gdzie ulegają oksydacji i fagocytozie przez makrofagi (rozdz. VII.2). U osób z ZKM nasilony jest katabolizm
apoA-I, co skutkuje spadkiem stężenia lipoproteiny HDL. Badania epidemiologiczne poka212
zują, że istnieje odwrotna zależność między stężeniem HDL a ryzykiem rozwoju miażdżycy
[25]. Na anty-aterogenne działanie HDL składają się [26]: (i) zwiększony zwrotny transport cholesterolu zgromadzonego w makrofagach, śródbłonku naczyniowym i komórkach
mięśni gładkich blaszek miażdżycowych do HDL i transport cholesterolu zgromadzonego
w HDL do watroby; (ii) osłabienie modyfikacji oksydacyjnej LDL – bezpośrednio przez
wymiatanie ROS przez apoA-I i pośrednio przez przenoszenie utlenionych fosfolipidów
z LDL na HDL; (iii) ochrona śródbłonka naczyniowego – w badaniach in vitro pokazano,
że HDL zwiększa ekspresję/aktywność eNOS i biodostępność NO oraz hamuje ekspresję
cząstek adhezyjnych VCAM-1, ICAM-1 i E-selektyny;
3. Otyłości i ZKM towarzyszą pro-aterogenne zmiany profilu adipokin i cytokin, w tym: (i)
wzrost poziomu TNFα, IL-1β, IL-6 i MCP-1; (ii) wzrost poziomu leptyny – o jej istotnym
udziale w rozwoju miażdżycy w otyłości świadczą wyniki eksperymentów na myszach pozbawionych genu leptyny (ob/ob). Pomimo rozwoju ogromnej otyłości i cukrzycy, u zwierząt
nie obserwowano zmian miażdżycowych, które pojawiały się po podaniu egzogennej leptyny
[16]; (iii) wzrost poziomu insuliny – może brać udział w rozwoju miażdżycy poprzez: stymulację śródbłonkowej produkcji ROS, nasilenie proliferacji i aktywacji monocytów i wzrost
produkcji przez nie prozapalnych cytokin oraz zwiększanie migracji i proliferacji mięśni gładkich [17]; (iv) wzrost poziomu rezystyny – badania kliniczne pokazują, że jej poziom wzrasta w CNS i koreluje ze stężeniem IL-6 i rozpuszczalnych receptorów typu 2 dla TNFα [18].
Rezystyna jest modulatorem odpowiedzi zapalnej i odpowiada za rozwój promiażdżycowego
fenotypu w śródbłonku naczyniowym (wzrost ekspresji VCAM-1 i MCP-1) [19]; (v) spadek
poziomu adiponektyny – jej poziom spada u pacjentów z ZKM i spadek ten koreluje z nasileniem procesu miażdżycowego [20] natomiast jej wysoki poziom koreluje z niskim ryzykiem
wystąpienia zawału serca [22]. W układach eksperymentalnych adiponektyna zwiększała
ekspresję/aktywność eNOS i produkcję NO, zmniejsza ekspresje cząstek adhezyjnych i adhezję monocytów do śródbłonka, zmniejsza aktywność NF-κB i NF-κB-zależną produkcję
cytokin, zmniejsza fagocytozę ox-LDL przez monocyty/makrofagi i powstawanie komórek
piankowych oraz zmniejsza proliferacje i migrację komórek mięśni gładkich naczyń [21] oraz
(vi) spadek poziomu apeliny – jej działanie przeciwmiażdżycowe sugerują głównie badania
laboratoryjne. Dla przykładu, w modelu eksperymentalnym miażdżycy wykazano jej działanie anty-rodnikowe i zwiększające biodostępność NO [23]. Pokazano, że mężczyzn z toczącym się procesem miażdżycowym, stężenie apeliny spada w krążeniu systemowym i równocześnie rośnie w naczyniach wieńcowych [24]. Istnieje spekulacja, że apelina wydzielana jest
miejscowo jako inhibitor procesu miażdżycowego.
X.7. Zaburzenia czynności skurczowej serca w zespole
kardiometabolicznym
U pacjentów z ZKM dochodzi do przerostu [27] i upośledzenie kurczliwości mięśnia
sercowego [28]. Przyczyną tych zmian mogą być (ryc. 10.5.):
1. Miażdżyca i nadciśnienie tętnicze;
2. Ekspozycja serca na wysokie stężenie wolnych kwasów tłuszczowych skutkująca tzw. lipotoksycznością. Wolne kwasy tłuszczowe wychwytywane są przez serce proporcjonalnie do
213
ich osoczowego stężenia. W ZKM dochodzi do zwiększonego napływu kwasów tłuszczowych do kardiomiocytów i następnie gromadzenia się w komórkach cząsteczek acyloCoA,
które stają się substratem do komórkowej syntezy dwuacylogliceroli, triacylogliceroli (triglicerydów) i ceramidów (rozdz. IV). Diacyloglicerole są aktywatorami kinazy białkowej C
(PKC). Natomiast ceramidy, poprzez aktywację NF-κB i kaspazy 3, nasilają apoptozę kardiomiocytów [29], a poprzez aktywację błonowych kanałów potasowych [30], skracają czas
trwania potencjałów czynnościowych, co skutkuje mniejszym dokomórkowym napływem
jonów Ca2+ i osłabieniem siły skurczu kardiomiocytów;
3. Hiperinsulinemia; Utrzymujące się wysokie stężenie insuliny aktywuje pro-przerostowe ścieżki sygnalizacyjne w miokardium zależne od: (i) kinazy Akt-1, której aktywacja prowadzi do
aktywacji genów pro-przerostowych i równoczesnego zahamowania degradacji białek [31];
Akt-1 może być pobudzana bezpośrednio przez insulinę i aktywację receptora insulinowego,
lub pośrednio przez aktywację układu współczulnego i receptorów β2-adrenergicznych [32];
(ii) kinaz aktywowanych miogenem (MAPK) – ERK i p38, o szerokim spektrum działania
pro-przerostowego, promującego przebudowę i pro-apoptycznego [33];
4. Zwiększona produkcja ROS z licznymi tego niekorzystnymi konsekwencjami [34-36];
5. Adipokiny:
a. Osoczowe stężenie leptyny koreluje dodatnio (i) z ciśnieniem tętniczym krwi u pacjentów z nadciśnieniem, niezależnie od masy ciała [37] oraz (ii) z grubością ściany LV serca,
niezależnie od ciśnienia tętniczego [38];
b. U ludzi niskie stężenie adiponektyny w osoczu jest związane z postępującym przerostem mięśnia sercowego i upośledzeniem funkcji rozkurczowej [39]. W badaniach eksperymentalnych, u myszy pozbawionych genu adiponektyny przeciążenie ciśnieniowe
prowadzi do zwiększonego przerostu i upośledzenia funkcji skurczowej LV oraz wzrostu
śmiertelności w porównaniu ze zwierzętami kontrolnymi. Zjawiskom tym zapobiega terapia genowa przywracająca ekspresję adiponektyny [40];
c. U mysz pozbawionych genu apeliny dochodzi do większego upośledzenia funkcji skurczowej serca związanego z wiekiem, a przeciążenie ciśnieniowe powoduje większy przerost
i upośledzenia funkcji skurczowej LV w porównaniu ze zwierzętami kontrolnymi [41].
Piśmiennictwo
1. Mottillo S, Filion KB, Genest J, et al. The metabolic syndrome and cardiovascular risk a systematic
review and meta-analysis. J Am Coll Cardiol. 2010; 56(14): 1113-32.
2. Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim
statement of the International Diabetes Federation Task Force on Epidemiology and Prevention;
National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation;
International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120(16):1640-5.
3. IDF Consensus Worldwide Definition of the Metabolic Syndrome. www.idf.org
4. Ford ES. Prevalence of the metabolic syndrome defined by the International Diabetes Federation
among adults in the U.S. Diabetes Care. 2005;28(11):2745-9.
5. Wyrzykowski B, Zdrojewski T, Sygnowska E i wsp. Epidemiologia zespołu metabolicznego w Polsce.
Wyniki programu WOBASZ. Kardiologia Polska 2005;63(6 Supl 4):S641-4.
214
6. Booth FW, Chakravarthy MV, Spangenburg EE. Exercise and gene expression: physiological regulation of the human genome through physical activity. JPhysiol. 2002;543(Pt 2):399-41.
7. Farooqi IS, Matarese G, Lord GM, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin
Invest. 2002; 110(8):1093-103.
8. Fasshauer M, Kralisch S, Klier M, et al. Adiponectin gene expression and secretion is inhibited by
interleukin-6 in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2003; 301(4): 1045-50.
9. Hopkins TA, Ouchi N, Shibata R, et al. Adiponectin actions in the cardiovascular system. Cardiovasc
Res. 2007; 74(1): 11-8.
10. Chen BH, Song Y, Ding EL, et al. Circulating levels of resistin and risk of type 2 diabetes in men and
women: results from two prospective cohorts. Diabetes Care. 2009; 32(2): 329-34.
11. Berndt J, Klöting N, Kralisch S, et al. Plasma visfatin concentrations and fat depot-specific mRNA
expression in humans. Diabetes. 2005; 54(10): 2911-6.
12. Fukuhara A, Matsuda M, Nishizawa M, et al. Visfatin: a protein secreted by visceral fat that mimics
the effects of insulin. Science. 2005; 307(5708): 426-30.
13. Tran TT, Yamamoto Y, Gesta S, et al. Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab. 2008; 7(5): 410-20.
14. Mark AL, Shaffer RA, Correia ML, Morgan DA, Sigmund CD, Haynes WG. Contrasting blood pressure effects of obesity in leptin-deficient ob/ob mice and agouti yellow obese mice. J Hypertens.
1999;17:1949–53.
15. Shek EW, Brands MW, Hall JE. Chronic leptin infusion increases arterial pressure. Hypertension.
1998;31:409–14.
16. Redon J, Cifkova R, Laurent S, et al. Mechanisms of hypertension in the cardiometabolic syndrome.
J Hypertens. 2009;27(3):441-51.
17. Hasty AH, Shimano H, Osuga J, et al. Severe hypercholesterolemia, hypertriglyceridemia, and
atherosclerosis in mice lacking both leptin and the low density lipoprotein receptor. J Biol Chem.
2001;276:37402–8.
18. Dubey L, Hesong Z. Role of leptin in atherogenesis. Exp Clin Cardiol. 2006;11(4):269-75.
19. Reilly MP, Lehrke M, Wolfe ML, et al. Resistin is an inflammatory marker of atherosclerosis in humans. Circulation. 2005;111(7):932-9.
20. Verma S, Li SH, Wang CH, et al. Resistin promotes endothelial cell activation: further evidence of
adipokine-endothelial interaction. Circulation. 2003; 108(6): 736-40.
21. Kumada M, Kihara S, Sumitsuji S, et al. Osaka CAD Study Group. Coronary artery disease. Association of hypoadiponectinemia with coronary artery disease in men. Arterioscler Thromb Vasc Biol.
2003;23(1):85-9.
22. Han SH, Sakuma I, Shin EK, et al. Antiatherosclerotic and anti-insulin resistance effects of adiponectin: basic and clinical studies. Prog Cardiovasc Dis. 2009;52(2):126-40.
23. Pischon T, Girman CJ, Hotamisligil GS, et al. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA. 2004; 291(14): 1730-7.
24. Pitkin SL, Maguire JJ, Kuc RE, et al. Modulation of the apelin/APJ system in heart failure and atherosclerosis in man. Br J Pharmacol. 2010; 160(7): 1785-95.
25. Gordon DJ, Probstfield JL, Garrison RJ, et al. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation. 1989;79(1):8-15.
26. Natarajan P, Ray KK, Cannon CP. High-density lipoprotein and coronary heart disease: current and
future therapies. J Am Coll Cardiol. 2010;55(13):1283-99.
27. de Simone G, Palmieri V, Bella JN, et al. Association of left ventricular hypertrophy with metabolic
risk factors: the HyperGEN study. J Hypertens. 2002; 20(2): 323-31.
215
28. de las Fuentes L, Brown AL, Mathews SJ, et al. Metabolic syndrome is associated with abnormal left
ventricular diastolic function independent of left ventricular mass. Eur Heart J. 2007; 28(5): 553-9.
29. Zhang DX, Fryer RM, Hsu AK, et al. Production and metabolism of ceramide in normal and ischemic-reperfused myocardium of rats. Basic Res Cardiol. 2001; 96(3): 267-74.
30. Liu GX, Hanley PJ, Ray J, et al. Long-chain acyl-coenzyme A esters and fatty acids directly link metabolism to K(ATP) channels in the heart. Circ Res. 2001; 88(9): 918-24.
31. Sharma N, Okere IC, Duda MK, et al. Potential impact of carbohydrate and fat intake on pathological left ventricular hypertrophy. Cardiovasc Res. 2007; 73(2): 257-68.
32. Morisco C, Condorelli G, Trimarco V, et al. Akt mediates the cross-talk between beta-adrenergic and
insulin receptors in neonatal cardiomyocytes. Circ Res. 2005; 96(2): 180-8.
33. Wang CC, Goalstone ML, Draznin B. Molecular mechanisms of insulin resistance that impact cardiovascular biology. Diabetes. 2004; 53(11): 2735-40.
34. Bidasee KR, Nallani K, Yu Y, et al. Chronic diabetes increases advanced glycation end products on
cardiac ryanodine receptors/calcium-release channels. Diabetes. 2003; 52(7): 1825-36.
35. Candido R, Forbes JM, Thomas MC, et al. A breaker of advanced glycation end products attenuates
diabetes-induced myocardial structural changes. Circ Res. 2003; 92(7): 785-92.
36. Wakasaki H, Koya D, Schoen FJ, et al. Targeted overexpression of protein kinase C β2 isoform in
myocardium causescardiomyopathy. Proc Natl Acad Sci U S A. 1997; 94(17): 9320–9325.
37. Khokhar KK, Sidhu S, Kaur G. Correlation between leptin level and hypertension in normal and
obese pre- and postmenopausal women. Eur J Endocrinol. 2010; 163(6): 873-8.
38. Paolisso G, Tagliamonte MR, Galderisi M et al. Plasma Leptin Level Is Associated With Myocardial
Wall Thickness in Hypertensive Insulin-Resistant Men. Hypertension 1999; 34: 1047-1052.
39. Hong SJ, Park CG, Seo HS, et al. Associations among plasma adiponectin, hypertension, left ventricular diastolic function and left ventricular mass index. Blood Press. 2004; 13(4): 236-42.
40. Shibata R, Ouchi N, Ito M, et al. Adiponectin-mediated modulation of hypertrophic signals in the
heart. Nat Med. 2004; 10(12): 1384-9.
41. Kuba K, Zhang L, Imai Y, et al. Impaired heart contractility in Apelin gene-deficient mice associated
with aging and pressure overload. Circ Res. 2007; 101(4): e32-42.
216
ISBN 978-83-62110-23-0
EGZEMPLARZ BEZPŁATNY
CZŁOWIEK – NAJLEPSZA INWESTYCJA!
Centrum Medyczne Kształcenia Podyplomowego w Warszawie
PATOFIZJOLOGIA MIAŻDŻYCY I CHOROBY NIEDOKRWIENNEJ SERCA
Projekt współfinansowany przez Unię Europejską z Europejskiego Funduszu Społecznego
„Kształcenie w ramach procesu specjalizacji lekarzy deficytowych specjalności
tj. onkologów, kardiologów i lekarzy medycyny pracy”
PATOFIZJOLOGIA
MIAŻDŻYCY
I CHOROBY
NIEDOKRWIENNEJ
SERCA
Redaktor naukowy
prof. dr hab. n. med. Andrzej Beręsewicz