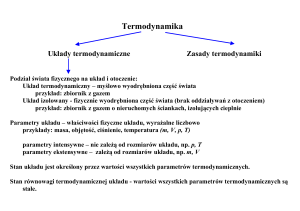
I zasada termodynamiki.
Jest to zasada zachowania energii w termodynamice - równoważność pracy i ciepła.
Rozważmy proces adiabatyczny sprężania gazu od V1 do V2:
V2
∫ dW
zew, ad
= U 2 − U 1 - wykonanie pracy powoduje przyrost energii wewnętrznej układu.
V1
We wzorze tym U1 jest początkową energią wewnętrzną gazu a U2 jego energią
końcową.
Jeśli uwzględnimy, że energię możemy dostarczać zarówno w postaci ciepła jak i pracy,
możemy napisać:
U 2 − U 1 = ∆U = ∆W zew + ∆Q zew
(1)
gdzie ∆Wzewn jest pracą wykonaną nad gazem przez siły zewnętrzne a ∆Qzew ciepłem
dostarczonym do gazu. Jeśli proces przebiega odwracalnie to od postaci całkowej
równania można przejść do postaci różniczkowej
(2)
dU = dWzew + dQzew
I zasada termodynamiki (zapis Clausiusa):
Jeśli pracę objętościową wyszczególnimy to można zapisać:
dU = dQ zew − pdV + ∑ X i dai
i
Symbol d używamy na oznaczenie formy Pfaffa, co zaznaczamy zazwyczaj przez „d”
kreślone.
Ciepło właściwe
Z doświadczenia wiemy, że na ogrzanie ciała od temperatury T1 do T2 potrzeba
proporcjonalnej do jego masy ilości ciepła:
∆Q ~ m(T2 − T1 )
1
∆Q = cm∆T
Wielkość c określoną następująco:
c=
1 ∆Q
m ∆T
nazywamy ciepłem właściwym.
Ilość dostarczonego ciepła, potrzebnego na zmianę temperatury ciała o ∆T, zależy
również od warunków w jakich ten proces następuje:
1 ∆Q
1 ∂Q
=
m ∆T X m ∂T X
gdzie X charakteryzuje typ przemiany - przemiana izobaryczna (X=p) lub izochoryczna
(X=V).
c X = lim
∆T → 0
Dalej będziemy zajmowali się ciepłem potrzebnym na ogrzanie 1 mola substancji o 1
stopień. Oznaczymy je przez „C”a jego wymiarem będzie:
J
[C] =
mol ⋅ K
Przykłady :
∂Q
CV =
- ciepło właściwe przy stałej objętości,
∂T V
∂Q
Cp =
∂T p
- ciepło właściwe przy stałym ciśnieniu.
Istnieje ogólny związek pomiędzy Cv i Cp dla dowolnej substancji i w dowolnym stanie
skupienia. Rozważmy energię wewnętrzną substancji jako funkcję U (T , V ) :
∂U
∂U
dU =
dT +
dV
(3)
∂T V
∂V T
Z I zasady termodynamiki mamy:
dQzew = dU + pdV
Wstawiając (3) do (4) dostajemy:
∂U
∂U
dQ zew =
dV + pdV
dT +
∂V T
∂T V
Jeśli dV = 0, to:
2
(4)
∂U
CV =
∂T V
Mamy więc :
∂U
/ : dT przy p = const
dQ zew = CV dT +
+ p dV
∂V T
∂U
∂V
∂Q
= CV +
+ p
∂T p
∂V T
∂T p
Ale lewa strona powyższego równania to po prostu Cp , stąd:
∂U
∂V
C p − CV =
+ p
∂V T
∂T p
Związek ten można uogólnić :
∂U
∂V
C X − CV =
+ p
∂V T
∂T X
Dla gazu doskonałego jego energia nie zależy od objętości:
∂U
=0
∂V T
a stąd wynika równanie Mayera :
C p − CV = R
Dla gazu van der Waalsa energia wewnętrzna U = U(T,V) zależy od temperatury i od
objętości. W tym wypadku dostajemy przybliżoną zależność:
2a
C p − CV ≈ R1 +
dla b << V
RTV
Dla gazu doskonałego wartości Cv i Cp wynoszą:
3
J
CV = R = 12,47
2
mol ⋅ K
J
5
C p = R = 20,97
2
mol ⋅ K
3
Równanie Mayera dobrze opisuje gazy szlachetne (jednoatomowe) a znacznie gorzej
substancje, które tworzą wieloatomowe cząsteczki.
Tabela przedstawia wartości Cp dla gazów rzeczywistych ( T=250C, p=po).
Gaz
Ar
Ne
N2
O2
H2
NH3
(amoniak)
C6H6
(benzen)
J
Cp
mol ⋅ k
20,79
20,79
29,12
29,36
29,83
R
2,5
2,5
3,5
3,53
3,59
35,67
4,29
81,73
9,83
Cp
Teoria ciepła właściwego (Zajmuje się tym fizyka ciała stałego).
- wiele wewnętrznych stopni swobody dostępnych dla bardziej skomplikowanych
cząstek. My będziemy zajmować się tym problemem w ramach fizyki statystycznej.
Zależność ciepła właściwego od temperatury:
4
Procesy izoparametryczne.
Idealizacja rzeczywiście przebiegających procesów oraz ich analiza jest pożyteczna dla
zrozumienia rzeczywistych przemian.
•
Proces izochoryczny (V = const):
dWukl = pdV = 0
dU = dQ zew = dQ zew jest różniczką zupełną
dU = CV dT
•
Proces izobaryczny (p = const):
V2
Wukl = p ∫ dV = p (V2 − V1 )
V1
Ponieważ ciśnienie jest stałe ( dp = 0 ), to możemy napisać:
dWukl = pdV = pdV + Vdp = d ( pV ) p
Z I zasady termodynamiki mamy :
(dQzew ) p
= dU + d ( pV ) p = d (U + pV ) p = (dH ) p
gdzie wielkość H = U + pV nazywamy entalpią układu.
∂H
∂Q
Cp =
⇒ Cp =
∂T p
∂T p
Entalpia jest funkcją stanu, której przyrost w procesie izobarycznym równa się
ciepłu dostarczonemu do układu.
•
Proces izotermiczny (T = const):
Z I zasady termodynamiki mamy :
dQ zew = CV dT + pdV
5
ale ponieważ temperatura jest stała, więc dT = 0 ⇒ dU = 0 ,
dQ zew = pdV = dWuk
Ciepło przekazane do układu zostaje zużyte na wykonanie pracy
Pracę dla procesu kwazistatycznego (dla gazu doskonałego, gdzie pV=RT) można
policzyć :
W =
V2
V2
V
dV
= RT ln 2
V
V1
V1
∫ pdV = RT ∫
V1
•
[policzyć dla gazu van der Waalsa]
Proces adiabatyczny
- nie występuje wymiana ciepła między układem i otoczeniem
- jest to także proces izoparametryczny
dU = dW zew = dW zew = − dWuk
Stąd dla gazu doskonałego:
CV dT + pdV = 0 ,
⇓
p
dV
dT = −
C
V
RT
p=
- gaz się rozszerza (V↑, T↓)
V
dT
R dV
+
=0
T CV V
Po scałkowaniu mamy:
ln T +
R
ln V = const
CV
Z równania Mayera:
Cp
R Cp
=
−1= γ −1 , γ =
CV CV
CV
⇓
γ −1
= const ⇒ TV γ −1 = const
ln TV
Korzystając ze związku: T =
pV
, dostajemy równanie adiabaty – równanie
R
Poissona:
pV γ = const
6
II zasada termodynamiki
I zasada termodynamiki nie daje wskazówek co do kierunku przebiegu procesów
termodynamicznych. Rudolf Clausius tak sformułował II zasadę termodynamiki:
Niemożliwy jest samorzutny przepływ ciepła od ciała mniej nagrzanego do ciała
gorętszego.
Wymuszony przepływ jest możliwy – maszyny chłodnicze
Ta sama zasada w sformułowaniu Kelvina:
Niemożliwe jest otrzymywanie pracy mechanicznej z jakiegokolwiek układu
materialnego przez oziębienie go poniżej temperatury najzimniejszego z
otaczających obiektów.
Sformułowanie Ostwalda (1901) :
Niemożliwe jest uzyskanie perpetuum mobile II rodzaju.
7