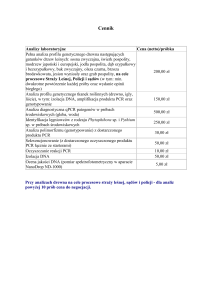
Uniwersytet Gdański, Wydział Biologii
Katedra Genetyki Molekularnej Bakterii
Katedra Biologii i Genetyki Medycznej
Katedra Biologii Molekularnej
Biologia i Biologia Medyczna II rok
Przedmiot:
Biologia Molekularna z Biotechnologią
===============================================================================================
Ćwiczenia 1
Klonowanie in silico
Klonowanie molekularne jest metodą inżynierii genetycznej wykorzystywaną do tworzenia
i powielania (klonować = tworzyć identyczne kopie) zrekombinowanych wektorów, niosących wybrany
fragment DNA. W praktyce laboratoryjnej klonowanie DNA polega na łączeniu (czyli rekombinacji)
cząsteczki wektora (plazmidu lub faga) z wybranym fragmentem DNA (wstawką) i wprowadzeniu takiego
tworu do organizmu, w którym uzyskany hybrydowy DNA może amplifikować się z wykorzystaniem
aparatu enzymatycznego gospodarza. W efekcie otrzymuje się klon, czyli zbiór cząsteczek identycznych pod
względem genetycznym. Narzędziami w klonowaniu są szczepy bakteryjne, wektory i enzymy. Pierwszym
etapem klonowania jest zwykle planowanie konstrukcji zrekombinowanego wektora in silico (ryc. 1 A).
Następnie z wykorzystaniem szeregu technik biologii molekularnej przeprowadza się syntezę
zaprojektowanych konstruktów in vitro (ryc. 1 B). Kolejnym etapem jest powielanie in vivo otrzymanych
uprzednio konstruktów (ryc 1 C). Stworzone w ten sposób nowe zrekombinowane cząsteczki DNA mogą
być wykorzystane do przeprowadzenia dowolnych eksperymentów.
A) Tworzenie zrekombinowanych wektorów in silico, czyli
planowanie konstrukcji zrekombinowanego wektora z
wykorzystaniem programów komputerowych.
In silico – (łac. „w krzemie”), prowadzenie eksperymentów/symulacji z
wykorzystaniem komputera.
B) Konstrukcja zrekombinowanych wektorów in vitro,
czyli tworzenie zaprojektowanych wektorów z
wykorzystaniem technik biologii molekularnej (np. PCR,
trawienie restrykcyjne) w warunkach laboratoryjnych.
In vitro – (łac. „w szkle”), prowadzenie badań poza organizmem
(„w probówce”).
WEKTOR
WSTAWKA
ZREKOMBINOWANY
WEKTOR
C) Powielenie skonstruowanych wektorów in vivo,
dzięki wykorzystaniu zdolności wektora do
autonomicznej replikacji wewnątrz komórek gospodarza.
In vivo – (łac. „na żywym”), prowadzenie badań wewnątrz żywego
organizmu.
Rycina 1. Uproszczony schemat przedstawiający etapy klonowania molekularnego.
1. Wektory genetyczne
Wektorami nazywamy elementy genetyczne służące do przenoszenia informacji genetycznej do
wnętrza komórek. Istotną cechą wektorów genetycznych jest ich zdolność do autonomicznej (tj. niezależnej
1
od chromosomu gospodarza) replikacji w komórkach gospodarza. Wektorami genetycznymi mogą być
plazmidy lub wirusy (w tym bakteriofagi), jednak w procesie klonowania najczęściej wykorzystuje się
wektory plazmidowe (ryc. 2). Plazmidy są cząsteczkami DNA występującymi naturalnie w komórkach
bakteryjnych, jednak na potrzeby inżynierii genetycznej najczęściej wykorzystuje się zmodyfikowane
pochodne naturalnych plazmidów. W celu uzyskania wektora plazmidowego użytecznego w procesie
klonowania DNA, plazmidy zwykle zmodyfikowane są w następujący sposób:
1. Masa cząsteczkowa plazmidu zostaje zredukowana. Obecność we wnętrzu komórki dużych ilości
dodatkowej informacji genetycznej kodowanej w plazmidowym DNA wiąże się z pogorszeniem kondycji
komórki (tzw. fitness), a nawet spadkiem jej żywotności. Rekombinacja wektora z klonowanym fragmentem
DNA powoduje z kolei wzrost masy cząsteczkowej plazmidu (i ilości informacji genetycznej zawartej
w plazmidowym DNA). Można zatem powiedzieć, że im mniejszy wektor, tym większa jego pojemność.
Oznacza to, że można wykorzystać go do przenoszenia do komórki większych fragmentów DNA bez ryzyka
znacznego obniżenia fitness komórek.
2. Geny warunkujące możliwość transferu zrekombinowanego plazmidu do innych bakterii zostają
usunięte. Dzięki temu przekazywanie wektora odbywać się będzie jedynie wertykalnie (czyli z komórek
macierzystych do potomnych).
3. Utworzenie miejsca wielokrotnego klonowania (ang. multiple cloning site– MCS). MCS tworzy się
zwykle poprzez integrację z plazmidem syntetycznego oligonukleotydu zawierającego sekwencje DNA
rozpoznawane przez wybrane enzymy restrykcyjne. Ułatwia to integrację z wektorem plazmidowym
dowolnego DNA ze względu na większy wybór enzymów restrykcyjnych, które można zastosować w tym
procesie.
4. Obecność jednego lub kilku genów markerowych (markerów selekcyjnych), służących do
wyróżnienia i selekcji transformantów (komórek, które pobrały zrekombinowany wektor) na podstawie ich
fenotypu.
Rycina 2. Schemat przedstawiający budowę wektora plazmidowego.
2. Wstawka
Wykorzystując technikę klonowania molekularnego można tworzyć i powielać plazmidy zawierające
dowolne fragmenty DNA (tzw. „wstawki” lub „inserty”). Jednym z najczęściej wykorzystywanych
sposobów przygotowania wstawki jest jej powielenie (amplifikacja) z wykorzystaniem techniki łańcuchowej
reakcji polimerazy (ang. polymerase chain reaction – PCR). W tym celu należy zaprojektować startery do
reakcji PCR, czyli krótkie oligonukleotydy, komplementarne do matrycowego DNA, który zamierzamy
powielić. Startery do amplifikacji wstawki projektuje się zwykle w taki sposób, aby możliwe było trawienie
restrykcyjne amplifikowanych fragmentów DNA, a następnie ich integracja z wektorem plazmidowym.
Dobrze zaprojektowane startery posiadają następujące cechy:
Długość startera: 19-25 nt
Temperatura topnienia: 58-62°C
Oba startery w parze powinny mieć zbliżoną temperaturę topnienia [Tm]
40< %GC < 60
2
Sekwencja startera powinna być pozbawiona powtórzeń (ang. runs), np. ‘AAAA’
Starter nie powinien mieć powtórzeń G/C na końcach 5’ i 3’, ale dobrze jeśli występuje jeden nt ‘C’
lub ‘G’
3. Enzymy
W procesie klonowania molekularnego często wykorzystuje się różnego rodzaju enzymy. Należą do
nich:
A) Enzymy restrykcyjne - izolowane z bakterii białka o aktywności endonukleaz, zdolne do
rozpoznawania specyficznych sekwencji w DNA i do przecinania dwuniciowej cząsteczki
DNA w określonym miejscu. Wskutek cięcia enzymami restrykcyjnymi, w DNA powstają
tępe lub lepkie końce, co umożliwia zaplanowanie integracji z wektorem plazmidowym
dowolnego DNA w wybranej orientacji.
B) Alkaliczna fosfataza – enzym, który katalizuje reakcję usuwania grup 5’ fosforanowych z
DNA, RNA oraz tri fosforanów. Defosforylacja końców DNA podczas przygotowywania
wektora do klonowania uniemożliwia jego recyrkularyzację („zamykanie się” pustych, nie
zawierających wstawki wektorów)
C) Fragment Klenowa polimerazy DNA I – białko będące produktem proteolizy polimerazy
DNA I z E. coli. Posiada aktywność polimerazy (3'→5’) i aktywność egzonukleolityczną
3'→5’, jednak bez aktywności egzonukleolityczną 5'→3’. Enzym ten ma zatem zdolność
katalizowania wiernej reakcji polimeryzacji bez degradacji końców 5’. W procesie
klonowania molekularnego enzym ten wykorzystywany jest w celu tworzenia tępych końców
DNA (poprzez „wypełnianie” lepkich końców – dobudowywanie nici DNA do końca 3’,
na matrycy końców DNA wysuniętych w kierunku 5’)
D) Ligazy DNA – są enzymami katalizującymi tworzenie wiązania fosfodiestrowego pomiędzy
dwoma fragmentami DNA. Funkcją ligaz DNA jest łączenie przerw w DNA, co ma miejsce
przy wszystkich procesach angażujących syntezę nowych nici DNA (replikacja – łącznie
fragmentów Okazaki, naprawa DNA) czy wycinanie i wstawianie ich fragmentów
(rekombinacja genetyczna).
Ligacja– proces łączenia wiązaniem fosfodiestrowym dwóch fragmentów lub końców nici
DNA za pomocą ligazy. Proces ligacji wymaga energii w postaci ATP lub NAD (w
zależności od rodzaju ligazy) (ryc. 3).
Rycina 3. Schemat procesu ligacji komplementarnych końców DNA wektora i wstawki.
4.
1.
2.
3.
Zagadnienia do samodzielnego przygotowania:
Rodzaje wektorów do klonowania
Wektory wahadłowe
Wektory wysoko- i niskokopijne oraz ich zastosowanie
3
5. Literatura
1. J. Kur „Podstawy inżynierii genetycznej. Teoria, ćwiczenia, testy.” Wydawnictwo PG, 1994.
2. B. Zalewska-Piątek, M. Olszewski, R. Piątek, S. Milewski, J. Kur „Biologia molekularna. Ćwiczenia
laboratoryjne.” Wydawnictwo PG, 2009.
Część praktyczna
Celem ćwiczenia jest zaplanowanie konstrukcji pochodnej plazmidu pUC18 zawierającej gen „λO” z
wykorzystaniem programu komputerowego Serial Cloner, czyli klonowanie in silico genu „λO” w wektorze
pUC18. Podczas kolejnych zajęć (ćwiczenia 2-4) przeprowadzone zostanie klonowanie molekularne z
wykorzystaniem technik inżynierii genetycznej w warunkach laboratoryjnych, zgodnie z projektem
opracowanym podczas ćwiczenia 1. Poniżej wymieniono etapy klonowania genu „λO” oraz zaznaczono, na
których ćwiczeniach dany etap zostanie wykonany (ryc. 4):
1. Izolacja wektora plazmidowego pUC18. Realizacja: ćwiczenie 2
2. Przygotowanie wstawki (fragment DNA zawierający gen „λO”) z wykorzystaniem reakcji PCR.
Realizacja: ćwiczenie 3
3. Trawienie restrykcyjne wektora oraz wstawki enzymami BamHI oraz PstI. Realizacja: ćwiczenie 3
4. Ligacja, czyli reakcja łączenia wektora ze wstawką. Realizacja: ćwiczenie 4
5. Wprowadzenie zrekombinowanego wektora plazmidowego (pochodna plazmidu pUC18 niosąca gen
„λO”) ) do komórek gospodarza, czyli transformacja komórek biorcy otrzymanym
zrekombinowanym plazmidem. Realizacja: ćwiczenie 4
6. Selekcja transformantów - po ćwiczeniu 4
Rycina 4. Schemat klonowania molekularnego z wykorzystaniem reakcji ligacji.
4
Wykorzystanie programu Serial Cloner podczas kolejnych etapów klonowania in silico:
1. Wektor plazmidowy pUC18 – wizualizacja sekwencji, MCS oraz dostępnych genów markerowych.
2. Przygotowanie wstawki pochodzącej z plazmidu pCB104 (gen λO) – symulacja przebiegu reakcji
PCR
3. Trawienie restrykcyjne wektora i wstawki (enzymami BamHI oraz PstI) – dobór enzymów
restrykcyjnych i przedstawienie graficzne powstałych fragmentów
4. Ligacja czyli reakcja łączenia wektora ze wstawką – wizualizacja powstałego zrekombinowanego
wektora plazmidowego
Wprowadzenie do programu:
Widok okna programu.
Podstawowe narzędzia, które będziemy wykorzystywać w pracy z tym programem na zajęciach:
1 – otwiera nową sekwencję do obróbki
2 – otwiera sekwencję DNA zapisaną na dysku, nad którą wcześniej pracowaliśmy
3 – zapisuje zmienioną sekwencję DNA
4 – pokazuje w sposób graficzny otwartą sekwencji DNA
5 – pokazuje szczegóły wczytanej sekwencji z miejscami restrykcyjnymi
6 – wskazuje ilość miejsc cięcia dla danego enzymu restrykcyjnego w wybranej sekwencji DNA
7 –pozwala przeprowadzić analizę po użyciu enzymów restrykcyjnych na żelu agarozwym
8 – zaznacza miejsca z genami , które warunkują specyficzne cechy wektora
9 – pozwala odnaleźć odpowiednie miejsca na wskazanej sekwencji, np. miejsca restrykcyjne
10 –pozwala na przeprowadzenie wirtualnej ligacji fragmentów DNA
11 – pozwala na projektowanie primerów i przeprowadzenie wirtualnej reakcji PCR
12 – pozwala na sprawdzenie miejsc homologicznych pomiędzy sekwencjami
5
Wprowadzanie sekwencji do programu:
file
open
wybór pliku (plazmid pCB104)
w celu wizualizacji plazmidu wybierz opcję „Graphic Map” , a następnie wybierz opcję
„Circularize”
Po wczytaniu danej sekwencji DNA do programu, możemy określić jej wielkość, wskazać miejsce inicjacji
replikacji, czy sprawdzić występujące tu markery selekcyjne.
Projektowanie starterów/ reakcja PCR:
Aby uzyskać produkt po reakcji PCR i zintegrować go z wektorem, konieczne jest zaprojektowanie
starterów komplementarnych do interesującej nas sekwencji DNA (matrycy DNA), którą chcemy powielić.
Podczas konstrukcji wprowadzamy miejsce cięcia dla enzymów restrykcyjnych na końcu 5’, aby możliwe
było trawienie restrykcyjne, a następnie reakcja ligacji.
6
Wprowadzamy sekwencję matrycową i zaznaczamy fragment sekwencji, który zamierzamy powielić.
Sprawdzamy czy starter ma odpowiednią długość, ilość par GC, temperaturę topnienia. Za pomocą tego
programu możemy również przeprowadzić wirtualną reakcję PCR z wykorzystaniem uprzednio
zaprojektowanych pimerów. Wczytujemy sekwencję matrycową (opcja u dołu okna „Run a PCR”), czyli tę
samą, którą wykorzystaliśmy do projektowania starterów, i otrzymujemy produkt PCR, który później
będziemy wykorzystać do reakcji ligacji.
Etapy:
wybierz opcję PCR
wstaw sekwencje rozpoznawane i cięte przez oba enzymy restrykcyjne na końcach 5’
projektowanych primerów w miejscu „oligo tail not indentical to the target sequence”, oraz
sekwencje pochodzące z plazmidu pCB104 w miejscu „segment identical to the target sequence” .
Po wyborze sekwencji pochodzącej z plazmidu pCB104 należy zaznaczyć opcję „ use as PCR
forward primer” oraz „ use as PCR reverse primer”
wstaw matrycę „select target DNA” i wybierz pCB104
wciśnij Run PCR
Wirtualna ligacja wektora i wstawki:
Program ten pozwala na przeprowadzenie wirtualnej reakcji ligacji dwóch fragmentów DNA: wektora oraz
wstawki (produktu reakcji PCR), które zostały potrawione tymi samymi enzymami restrykcyjnymi.
Wprowadzamy wcześniej przygotowaną i sprawdzoną sekwencję DNA do programu SerialCloner.
Zaznaczamy wybrane przez nas enzymy restrykcyjne i uzyskujemy informację na temat rodzaju cięcia
restrykcyjnego (końce tępe lub lepkie oraz wielkości otrzymanego fragmentu DNA).
7
Tak uzyskane fragmenty DNA ligujemy, a uzyskany zrekombinowany plazmid zapisujemy – poniżej mapa
powstałego konstruktu oraz dwóch plazmidów wykorzystanych do jego stworzenia.
Etapy ligacji:
po pocięciu enzymami restrykcyjnymi plazmidu pUC19 oraz wstawki z plazmidu pCB104
wybieramy opcję „Construct”, a następnie wstawiamy „otwartą” sekwencję nukleotydową plazmidu
pUC19 w „Select DNA 1” oraz sekwencję nukleotydową wstawki w „ Select DNA2”
reakcję wirtualnej ligacji przeprowadzamy poprzez naciśnięcie opcji „LIGATE”
Rycina 5. Schemat plazmindu pCB104 zawierającego gen λO, który zostanie wklonowany w MCS plazmidu pUC19
8
Rycina 6. Schemat plazmidu pUC19 zawierającego fragment genu lacZ kodujący N-końcowy fragment β-galaktozydazy
Rycina 7 Zrekombinowany plazmid uzyskany w wyniku klonowania in silico.
9