Ewa Maria Maunowicz – Nieklasyczna postać wrodzonego przerostu kory nadnerczyz powodu niedoboru 21-hydroksylazy (nk-wpn) u dziewczynek...
Vol. 7/2008 Nr 1(22)
Endokrynologia Pediatryczna
Pediatric Endocrinology
Nieklasyczna postać wrodzonego przerostu kory nadnerczy z powodu
niedoboru 21-hydroksylazy (nk-wpn) u dziewczynek i kobiet z objawami
androgenizacji
Nonclassic Congenital Adrenal Hyperplasia Due to 21-hydroxylase Deficiency
(NC-CAH) in Girls and Women with Androgenization Symptoms
Ewa Maria Małunowicz
Pracownia Zaburzeń Metabolizmu, Instytut-Pomnik Centrum Zdrowia Dziecka
Adres do korespondencji: Instytut-Pomnik Centrum Zdrowia Dziecka, Pracownia Zaburzeń Metabolizmu, Al. Dzieci Polskich 20, 04-730 Warszawa,
e-mail: [email protected]
Słowa kluczowe:
Key words:
nieklasyczna postać wrodzonego przerostu kory nadnerczy; niedobór 21-hydroksylazy; androgenizacja kobiet; steroidy
nadnerczowe.
nonclassic adrenal hyperplasia; 21-hydroxylase deficiency; female androgenization; adrenal steroids
STRESZCZENIE/ABSTRACT
Wstęp: Nk-wpn jest w chwili obecnej coraz częściej rozpoznawany u dzieci z przedwczesnym pubarche oraz
nastolatek i kobiet z objawami androgenizacji. Objawy kliniczne są bardzo różnorodne i steroidogeneza nadnerczowa
nie jest jeszcze w pełni poznana. Celem pracy jest retrospektywna ocena objawów klinicznych oraz steroidogenezy
nadnerczowej u kobiet z nk-wpn. Pacjenci i metoda: 52 pacjentki podzielono na 3 grupy wiekowe: grupa I: wiek
2.5-9 lat (n=24); grupa II: wiek 11-17 lat (n=16); grupa III: wiek 20-37 lat (n=11) oraz 1 kobieta w wieku 62 lat.
U wszystkich pacjentek wykonano dobowy profil steroidowy metodą GC/MS. Wyniki: U pacjentek w grupie
I potwierdzono następujące objawy kliniczne: przedwczesne pubarche (95.8%), clitoromegalię (25%), trądzik
(16.7%); w grupie II: hirsutyzm (75%), clitoromegalię (12.5%), trądzik (12.5%), pierwotny brak miesiączki (12.5%),
oligomenorrhea (12.5 %), PCO (6.25%); w grupie III: hirsutyzm (90.9%), trądzik (18.8%), oligomenorrhea (36.4%),
PCO (18.2%), bezpłodność (9.2 %). U najstarszej, asymptomatycznej kobiety wykryto gruczolaki w nadnerczach.
Dobowe wydalanie metabolitów kortyzolu (F) było prawidłowe u wszystkich pacjentek, ale u 34.1% potwierdzono
zaburzenia jego metabolizmu - zmniejszoną aktywność 11β-HSD1. Dobowe wydalanie metabolitów F wykazało
dodatnią, istotną (p<0.001) korelację z wydalaniem metabolitów 17-OHP, 21-dezoksykortyzolu oraz androstendionu.
Dobowe wydalanie metabolitu aldosteronu (THAldo) było istotnie podwyższone u pacjentek w wieku 2.5-17 lat
w porównaniu do grup kontrolnych zdrowych dziewczynek. Wnioski: Objawy androgenizacji u kobiet z nk-wpn
nie są specyficzne. Łagodny defekt syntezy F, jak i również mineralokortykoidów, jest kompensowany zwiększoną
odpowiedzią nadnerczy do wytwarzania F i aldosteronu.
9
Praca oryginalna
Endokrynol. Ped., 7/2008;1(22):9-18
Introduction: NC-CAH has been increasingly recognized in children with precocious pubarche and adolescent or
adult hyperandrogenic women. Patients present a wide spectrum of clinical symptoms. Adrenal steroidogenesis has
not been fully explained. Aim: Retrospective evaluation of clinical symptoms and adrenal steroidogenesis in NCCAH females. Patients and method: 52 females were divided into 3 groups, according to the age group: group I,
age 2.5-9 yrs (n=24); group II, age 11-17 yrs (n=16); group III, age 20-37 yrs (n=11) and additionally 1 woman aged
62 yrs. In all patients 24 hrs urinary steroid profile by GC/MS was performed. Results: Patients in group I showed
the following symptoms: precocious pubarche (95.8%), clitoromegaly (25%), acne (16.7%); in group II: hirsutism
(75%), clitoromegaly (12.5%), acne (12.5%), primary amenorrhea (12.5), oligomenorrhea (12.5%), PCOS (6.25%);
in group 3: hirsutism (90.9%), acne (18.8%), oligomenorrhea (36.4%), PCOS (18.2%), infertility (9.1%). In the oldest,
asymptomatic woman adrenal incidentaloma was observed. Daily excretion of cortisol metabolites was normal in
all patients, but in 31.4% the diminished activity of 11β-HSD1 was found. Daily excretion of cortisol metabolites
correlated positively, significantly (p<0.001) with excretion of 17-hydroksyprogesterone, 21-desoxycortisol and
androstenedione metabolites. Daily excretion of aldosterone metabolite (THAldo) was significantly higher in girls
aged 2.5-17 yrs in comparison to age-matched control groups of healthy girls. Conclusions: The androgenization
symptoms in NC-CAH females are not specific. In NC-CAH patients glucorticoids as well as mineralocorticoids
synthesis mild defect is compensated by the increased adrenal response to produce cortisol and aldosterone. Pediatr.
Endocrinol., 7/2008;1(22):9-18
Wstep
Nieklasyczna postać wrodzonego przerostu kory
nadnerczy z niedoborem 21-hydroksylazy (nkwpn) jest jedną z najczęstszych chorób dziedziczonych autosomalnie recesywnie. Występuje u około
0.3 % białej populacji, jednak z różną częstotliwością, zależną od grupy etnicznej. U kobiet z objawami hyperandrogenizmu, nk-wpn można potwierdzić u 1% - 10% pacjentek [1-5]. Wstępne badania
wykonane w Polsce w latach poprzednich, potwierdziły występowanie nk-wpn u 8.4% dzieci z przedwczesnym pubarche oraz u 4.7% dziewcząt w okresie pokwitania z objawami androgenizacji. Ta częstość występowania nk-wpn u dzieci z przedwczesnym pubarche, była zbliżona do danych, pochodzących z innych krajów [6]. Pacjenci z nk-wpn,
w odróżnieniu od klasycznej postaci wpn, nie mają
objawów po urodzeniu. Objawy androgenizacji, pojedyncze lub złożone, pojawiają się najczęściej w
okresie około pokwitania jako przedwczesne pubarche u obu płci, trądzik, przyśpieszona szybkość
wzrastania i zaawansowanie wieku kostnego, wpływające na obniżenie ostatecznego wzrostu. U kobiet oprócz hirsutyzmu, który może być jedynym
objawem androgenizacji, bardzo często pojawia się
dysfunkcja jajników i zaburzenia miesiączkowania
(amenorrhea, cykle bezowulacyjne, oligomenorrhea), niepłodność. Sugeruje się, że jest to związane
ze zwiększoną konwersją nadmiaru androgenów do
estrogenów, które zaburzają wydzielanie gonadotropin. U niektórych kobiet pojawia się zespół poli10
cystycznych jajników. U starszych pacjentek może
się pojawić łysienie typu męskiego. [5 i piśmiennictwo cytowane, 7]. U niektórych pacjentek, pomimo
zaburzeń biochemicznych, objawy androgenizacji
mogą być nieobecne, ale pojawią się w okresie późniejszym [8].
Czasem u pacjentek można potwierdzić lekki
przerost łechtaczki, ale nie jest to nigdy obojnactwo
rzekomo męskie [6, 7]. Radiologiczne badania nadnerczy potwierdzają u około 40% pacjentek przerost kory nadnerczy lub gruczolaki jako efekt subtelnej, ale przewlekłej stymulacji nadnerczy przez
ACTH [9,10]. Rozpiętość wieku diagnozy nk-wpn
waha się od 6 miesięcy (przedwczesne pubarche) do
88 lat (incidentaloma w nadnerczach) [11,12].
Badania molekularne genu CYP21 potwierdziły
odrębny genotyp nk-wpn w porównaniu do klasycznej postaci wpn. Pacjenci z nk-wpn mogą posiadać
2 łagodne mutacje na obu allelach albo być złożonymi heterozygotami, z łagodną mutacją na jednym
allelu oraz ciężką na drugim allelu. U pacjentów z
nk-wpn badania in vitro ekspresji genu potwierdziły
zachowanie od 20 do 60% aktywności 21-hydroksylazy. U 98-99% pacjentów z nk-wpn istnieje korelacja fenotypu z genotypem [5,13].
Najlepszym testem diagnostycznym dla rozpoznania nk-wpn jest krótki test z ACTH (1-24), 0.25
mg, i.v, wykonany o godz. 7.30-9.30 rano, w fazie
folikularnej (3-8 dzień cyklu), z oznaczeniem stężenia 17-OHP w warunkach podstawowych i 60 minut po podaniu ACTH. Wartości 17-OHP po stymulacji powyżej 15 ng/ml (45.4 nmol/l) są pewne
diagnostyczne dla rozpoznania nk-wpn, a wartości
Ewa Maria Maunowicz – Nieklasyczna postać wrodzonego przerostu kory nadnerczyz powodu niedoboru 21-hydroksylazy (nk-wpn) u dziewczynek...
poniżej 10 ng/ml (30.3 nmol/l) wykluczają nk-wpn.
W zakresie 10-15 ng/ml istnieje pewien zakres
niepewności, czy nie są to wyniki uzyskane u
heterozygot wpn. Niezbędne jest wtedy wykonanie
badań genetycznych [14, 15].
Podstawowy poziom 17-OHP w surowicy, oznaczony przed godz. 8.00 rano (u miesiączkujących
kobiet w fazie folikularnej), może służyć jako badanie przesiewowe w kierunku rozpoznania nk-wpn,
aby nie wykonywać zbędnego testu z ACTH. Stężenie u dzieci poniżej 0.8 ng/ml (2.5 nmol/l), a u kobiet poniżej 2 ng/ml (6.0 nmol/l), wyklucza nk-wpn
[15]. Według Dewailly należy wykonać test stymulacji z ACTH wyłącznie u kobiet, u których uzyskano poranne wartości stężenia 17-OHP pomiędzy 2
a 4 ng/ml (6-12 nmol/l); wartości poniżej i powyżej tego zakresu mają już ujemną i pozytywną wartość diagnostyczną dla rozpoznanie nk-wpn [16].
Jednakże u 10% badanych 220 pacjentek z nk-wpn
stwierdzono podstawowe stężenia 17-OHP poniżej
2 ng/ml [7].
Alternatywną metodą diagnostyczną jest wykonanie profilu steroidowego w moczu, gdzie ocenia
się dobowe wydalanie metabolitów 17-OHP (pochodzenie nadnerczowo-gonadowe), 21-dezoksykortyzolu (pochodzenie wyłącznie nadnerczowe,
specyficzny marker niedoboru 21-hydroksylazy),
metabolitów kortyzolu oraz ich wzajemne stosunki, oceniające stosunek prekursorów do substratu
[6, 17, 18].
Celem pracy jest charakterystyka kliniczna i biochemiczna grupy 52 pacjentek z nk-wpn, zdiagnozowanych w jednym ośrodku, gdzie oceniono dobowe wydalanie metabolitów kortyzolu, 17-OHP,
21-dezoksykortyzolu, androstendionu oraz po raz
pierwszy w piśmiennictwie, także metabolitu aldosteronu THAldo i ich wzajemne korelacje.
Materiał i metody
Przeanalizowano retrospektywnie dane biochemiczne i kliniczne 52 dziewczynek i kobiet w wieku od 2.5 lat do 37 lat oraz dodatkowo jednej kobiety w wieku 62 lat, u których rozpoznano nk-wpn.
Pacjentki podzielono na 3 grupy wiekowe: grupa I
(n=24): wiek 2.5-9 lat (średnia wieku 6.6 lat); grupa
II (n=16): wiek 11-17 lat (średnia wieku 15.1 lat);
grupa III (n=11): wiek 20-37 lat (średnia 27.7 lat).
Były to pacjentki Poradni Endokrynologicznej IPCZD, jak również z terenu całej Polski, skierowane w latach 2000-2007 na wykonanie profilu stero-
idowego w moczu z powodu podejrzenia nk-wpn.
U 14 pacjentek wykonane uprzednio oznaczenia
w warunkach podstawowych stężenia 17-hydroksyprogesteronu (17-OHP) w surowicy, wykazały
ich podwyższony poziom (2.08-20.0 ng/ml, średnia
14.4 ng/ml) i były skierowane do weryfikacji wstępnego rozpoznania nk-wpn. U miesiączkujących pacjentek dobowa zbiórka moczu była wykonana pomiędzy 7 a 9 dniem cyklu. Podczas wykonywania
zbiórki moczu dieta pacjentek była bez kontroli podaży soli. Pięćdziesiąt pacjentek miało prawidłową
masę ciała, u dwóch stwierdzono otyłość. Objawy
kliniczne wszystkich pacjentek przedstawiono w tabeli I. Grupy kontrolne stanowiły: 24 dziewczynki w wieku 4-9 lat, 32 dziewczęta w wieku 13-18
lat oraz 11 kobiet w wieku 20-37 lat z prawidłową
masa ciała, bez stwierdzonych endokrynopatii.
U wszystkich pacjentek był wykonany profil
steroidowy w moczu metodą GC/MS wg procedury Skackletona [19]. Analizy wykonano na chromatografie gazowym z detektorem masowym HP
GC5890/MS6930, z 12.5 m kolumną kapilarną HP1 Ultra.
Analiza statystyczna:
Do opracowań statystycznych wyników wykorzystano funkcje statystyczne pakietu obliczeniowego Excell. Z powodu nieparametrycznego rozkładu wyniki przedstawiono jako mediana i zakres. W
analizie statystycznej do porównania wyników w
grupach zastosowano test U Manna-Whitneya. Do
oceny korelacji liniowej użyto współczynnika Pearsona. Za istotne statystycznie uznano p<0.05. W
obliczeniach statystycznych nie uwzględniono wyników 62-letniej pacjentki.
Wyniki
Dane kliniczne
Rozpiętość wieku diagnozy nk-wpn u badanych
pacjentek wahała się od 2.5 do 62 lat. Głównym objawem androgenizacji w grupie najmłodszych dzieci w wieku 2.5-9 lat (n=24) było przedwczesne pubarche (95.8%), clitoromegalia (25%) oraz trądzik
(16.7%). Wiek kostny był przyspieszony o 1-2 lata.
W grupie wiekowej 11-17 lat (n=16) głównym objawem był hirsutyzm (75%), ale u 31.2 % pacjentek pojawiły się zaburzenia funkcji jajników, takie
jak pierwotny brak miesiączki w wieku 17 lat (2 pacjentki), oligomenorrhea oraz zespół policystycznych jajników. U 12.5% pacjentek obserwowano
także clitoromegalię oraz u 12.5% trądzik. U jednej
11
Praca oryginalna
Endokrynol. Ped., 7/2008;1(22):9-18
pacjentki był on jedynym objawem androgenizacji.
U dorosłych kobiet w wieku 20-37 lat już u 90.9%
obserwowano hirsutyzm, a łącznie u 45.4% pacjentek także zaburzenia funkcji jajników (oligomenorrhea, PCOS, bezpłodność). Dwie pacjentki miały w
wywiadzie przedwczesne pubarche. U najstarszej
62-letniej asymptomatycznej kobiety wykryto jako
incidentaloma gruczolaki nadnerczy Zbiorcze zestawienie objawów klinicznych pacjentek z nk-wpn
przedstawiono w tab. I.
aktywność dehydrogenazy 11β-hydroksysteroidowej (11β-HSD1), powodującą zmniejszony dostęp
wytworzonego kortyzolu do receptora glikokortykoidowego (tab. III).
Dobowe wydalanie metabolitu aldosteronu tzn. tetrahydroaldosteronu (THAldo) było
istotnie zwiększone u pacjentek w wieku 2.5-9
lat (p=0.006), jak również w wieku 11-17 lat
(p=0.001), w porównaniu do wartości uzyskanych
w grupach kontrolnych (Ryc.1, Tab. II). U doro-
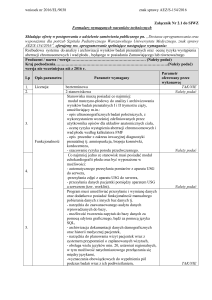
Tab. I. Objawy kliniczne u dziewcząt: kobiet z nk-wpn
Tab. I. Presenting clinical symptoms in girls and women with NC-CAH.
Wiek
Objawy
2.5-9 lat (n=24)
Liczba
%
11-17 lat (n=16)
Liczba
%
20-37 lat (n=11)
Liczba
Przedwczesne pubarche
23
95.8
-
Powiększenie łechtaczki
6
25
2
12.5
-
Trądzik
4
16.7
2
12,5
2
18.2
Hirsutyzm
1
4.2
12
75
10
90.9
Oligomenorrhea
-
2
12.5
4
36.4
Pierwotny brak miesiączki
-
1
6.2
-
PCO
-
1
6.2
2
18.2
-
Bezpłodność
-
-
1
9.1
-
Gruczolaki nadnerczy
Dane biochemiczne
W tab. II przedstawiono wyniki dobowego wydalania metabolitów steroidów nadnerczowych
pochodzących od 17-hydroksyprogesteronu (17OHP), 21-dezoksykortyzolu (21-DF), kortyzolu
(F), androstendionu oraz aldosteronu.
U wszystkich pacjentek, niezależnie od wieku, stwierdzono podwyższone wydalanie metabolitów 17-OHP (pregnantriol+17α-hydroksypregnanolon), 21-DF (pregnantriolon) oraz androstendionu (11-hydroksyandrosteron – jeden z jego głównych metabolitów), największe u dziewcząt w okresie pokwitania (11-17 lat).
Dobowe wydalanie skoniugowanych metabolitów F (THE+THF+allo-THF) było w normie u 48
pacjentek, a u 3 było nawet podwyższone (u dwóch
pacjentek z grupy I oraz jednej z grupy II). Pomimo
prawidłowego dobowego wytwarzania kortyzolu,
potwierdzono łącznie u 31.4% pacjentek zaburzony obwodowy metabolizm kortyzolu, tzn. obniżoną
12
2
%
-
18.2
62 lata
(n=1)
-
-
1
słych kobiet różnice te nie były istotne statystycznie. Najwięcej podwyższonych wyników dobowego wydalania THAldo obserwowano u pacjentek w okresie dojrzewania. Łącznie u 18 pacjentek
(34.6%) potwierdzono podwyższone, poza zakresem wartości referencyjnych, wydalanie THAldo,
a u trzech (5.9%) - obniżone.
Stosunek ilości wydalanych w ciągu doby metabolitów 17-OHP oraz 21-DF do metabolitów F,
oceniający pośrednio niedobór 21-hydroksylazy,
przedstawiono w tab. III.
Oceniony w ten sposób niedobór 21-hydroksylazy, nie wykazał istotnych różnic w trzech grupach wiekowych pacjentek, jeżeli wzięto pod uwagę stosunek sumy metabolitów 17-OHP (pochodzenie nadnerczowo-gonadowe) do sumy metabolitów kortyzolu. Natomiast potwierdzono istotną
statystycznie (p<0.02) ujemną korelację niedoboru
21-hydroksylazy z wiekiem rozpoznania nk-wpn,
ocenioną jako stosunek metabolitów 21-DF (po-
mediana
zakres
mediana
zakres
mediana
zakres
mediana
zakres
11-17 lat
(n=16)
Norma
11-17 lat
(n=32)
20-37 lat
(n=11)
Norma
20-37lat
(n=11)
2579,1
481,3
(179,4-991,8)
2705.6
1135,5-12400
489,5
(216,5-915,3)
3649.5
1960-28888,2
801,3
119,6
(163-279)
1540.2
811,3-4248
93,9
(10,3-195,3)
2057.4
940,6-26295
23,3
(8,4-82,7)
131,2
6,1
(3,5-49,8)
706.4
566-2370
9,8
(3,2-30,7)
854.0
510-7651,1
5,3
(2,0-56,7)
598.4
71,4-1982,4
Metabolit
21-DF:
PTN
5408,9
2267,3
994,5
(495-1633,4)
1245.0
648,4-2267,3
2551.9
1911,93641,8
1868,8
(1126- 3995,7)
849,4
(317-2060,2)
937.05
515-2802,1
353,4
(155,7-932,5)
303.0
40-778,5
2174,2
(1106- 4672,3)
2860.5
1854,8- 11022
1280,1
(112,72154,7)
1165.2
291,4- 3372,2
Metabolity F:
THE
THF
Skróty: 17-OHP = 17α-hydroksyprogesteron; 17-OHPN = 17α-hydroksypregnanolon; PT = pregnantriol; PTN = pregnantriolon
21-DF = 21-dezoksykortyzol; F = kortyzol; THE-tetrahydrokortyzon; THF = tetrahydrokortyzol
THAldo = tetrahydroaldosteron
(n=1)
62 lata
mediana
zakres
Norma
4-9 lat
(n=22)
93,9
(30-238,9)
1381.1
678-5298,8
mediana
Zakres
2.5-9 lat
(n=24)
672.5
310-2209
Metabolity 17-OHP:
PT
17-OHPN
Grupa
wiekowa
Tab. II. Wydalanie metabolitów steroidowych (μg/24 h) w 3 grupach wiekowych kobiet z nk-wpn
Ta. II. Daily excretion (μg/24h) of steroid metabolites in 3 age groups of women with NC-CAH
718,5
980,5
(288,52563,6)
700.0
374-3401,8
1058,7
(418-2662,8)
1198.3
547,4- 3655,7
692,6
(110,21381,5)
530.3
50,1- 1066,7
allo-THF
99,7
22,4
(7-53,3)
43.6
3.5-142,7
21
(7,0-58)
47.4
4,0-280,4
12,1
(6-31,6)
32.8
3,5-92,0
Metabolit
Aldo:
THAldo
3379,9
586,7
(113.9-788,9)
1980.9
996-3884,0
495,3
(105,9- 870)
2894.3
872-12450,9
118,4
(26-245)
754.2
207,7-1250
Metabolit
androstendionu:
11-OHAN
Ewa Maria Maunowicz – Nieklasyczna postać wrodzonego przerostu kory nadnerczyz powodu niedoboru 21-hydroksylazy (nk-wpn) u dziewczynek...
13
Praca oryginalna
Endokrynol. Ped., 7/2008;1(22):9-18
Tab. III. Ocena niedoboru aktywności 21-hydroksylazy i dehydrogenazy 11β-hydroksysteroidowej typu1 (11β-HSD1) w 3 grupach
wiekowych kobiet z nk-wpn
Tab. III. Evaluation of 21-hydroxylase and 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) deficiency in 3 age groups of
21-hydroxylase-deficient NC-CAH women
Metab.17-OHP/ metab. F*
mediana
(zakres)
Metab.21 DF**/
met.F x 100
mediana
(zakres)
Aktywność
11β-HSD1***
mediana
(zakres)
Liczba pacjentek
z obniżoną
aktywnością
11β-HSD1
2.5-9 lat
(n=24)
1.26
(0.37-4.21)
23
(3-73)
0.63
(0.09-1.28)
9 (37.5%)
Norma
(n=22)
0.057±0.019 (SD)
0. 24
(0.1-1.2)
0.86±0.20 (SD)
1.25
(0,81-5.05)
19
(8-58)
0.77
(0.2-2.06)
0.16±0.07(SD)
0.24
(0.1-1.5)
0.88±0.17(SD)
1.19
(0.40-5.36)
16
(6-54)
0.72
(0.44-1.61)
4 (36.4%)
0.17 ±0.1
0.15
(0.084-0.24)
1.04±0.18(SD)
Łącznie 16
(31.4%)
Grupa wiekowa
11-17 lat
(n=16)
Norma
(n=34)
20-37 lat
(n=11)
Norma
(n=11)
3 (17.7%)
* Metabolity 17-OHP: pregnantriol + 17α-hydroksypregnanolon
Metabolity kortyzolu (F): THE+THF+allo-THF
** Metabolit 21-dezoksykortyzolu (21-DF) = pregnantriolon
*** aktywność 11β-HSD1 = stosunek tetrahydrometabolitów kortyzolu do tetrahydro-kortyzonu (THF+allo-THF)/THE
Ryc. 1. Indywidualne wyniki dobowego wydania THAIdo w 3
grupach wiekowych 52 kobiet z nk-wpn w porównaniu do grup
kontorlnych. Grupy:
1–nk-wpn, wiek 2.5-9 lat (n=24); 2–grupa kontrolna, wiek 4-9 lat (n=24)
2–nk-wpn, wiek 11-17 lat (n=16); 4–grupa kontrolna, wiek 13-18 lat (n=32)
5–nk-wpn, wiek 20-37 lat (n=11); 6–grupa kontrolna, wiek 20-37 lat (n=11)
–––mediana w danej grupie wiekowej,
S=różnica istotna; NS–nieistotna
Fig. 1. Scattergram of daily excretion values of THAIdo in 3
age groups of 52 women with NC-CAH in comparison to control groups. Groups:
1–NC-CAH, aged 2.5-9 yrs (n=24); 2–healthy controls aged 4-9 yrs (n=24)
2–NC-CAH, aged 11-17 yrs (n=16); 4–healthy controls aged 13-18 yrs (n=32)
5–NC-CAH, aged 20-37 yrs (n=11); 6–healthy controls aged 20-37 yrs (n=11)
–––median in each age group,
S=significant difference; NS–no significant
14
chodzenie wyłącznie nadnerczowe) do kortyzolu.
Analiza statystyczna otrzymanych wyników wykazała:
1. Istotną (p<0.001) dodatnią korelację ilości wydalanych metabolitów kortyzolu i metabolitów
17-OHP (ryc.2),
2. Istotną (p<0.001) dodatnią korelację ilości wydalanych metabolitów kortyzolu i 21-DF (ryc.3),
3. Istotną (p<0.001) dodatnią korelację ilości wydalanych metabolitów kortyzolu i androstendionu (ryc.4),
4. Nie znaleziono istotnej korelacji pomiędzy ilością wydalanego metabolitu aldosteronu THAldo, a odpowiednio ilościami wydalanych metabolitów 17-OHP, 21-DF oraz F.
Dyskusja
Objawy kliniczne androgenizacji u 52 pacjentek
w wieku 2.5-62 lat były bardzo różnorodne, ale niespecyficzne dla rozpoznania nk-wpn (tab.I). Zarówno u dziewczynek z przedwczesnym pubarche, jak i
dziewcząt w okresie dojrzewania i kobiet z objawami androgenizacji, zaburzeniami funkcji jajników,
a także tylko incindentaloma w nadnerczach, nale-
Ewa Maria Maunowicz – Nieklasyczna postać wrodzonego przerostu kory nadnerczyz powodu niedoboru 21-hydroksylazy (nk-wpn) u dziewczynek...
ży wziąć pod uwagę nk-wpn w diagnostyce różnicowej przyczyn androgenizacji. Uzyskana charakterystyka klinicznych objawów pacjentek z nk-wpn
w zależności od ich wieku, jest zgodna z wynikami uzyskanymi z retrospektywnego podsumowania
danych klinicznych 220 pacjentek z nk-wpn, pochodzących z wielu ośrodków na świecie [7]. Uzyskane wyniki potwierdzają również w tej serii badań, że nk-wpn jest progresywną chorobą, z narastaniem ilości i ciężkości objawów androgenizacji
wraz z wiekiem, kiedy się obserwuje większy procent pacjentek z objawami hirsutyzmu, zaburzeniami funkcji gonad. Ważną sprawą jest wczesna diagnostyka nk-wpn, bo prawidłowe leczenie glikortykoidami cofa objawy androgenizacji [5].
Analizowany biochemiczny profil 24-godzinnej
funkcji nadnerczy u pacjentek z nk-wpn, potwierdził niektóre jego nowe aspekty, dotąd nieopisane
w piśmiennictwie. Jest sprawą znaną, że generalnie
u pacjentów z nk-wpn nie stwierdza się zwiększonego poziomu ACTH oraz niedoboru F. Podstawowy poziom F jest w normie, jak i również wolny F
w moczu [20-22]. U niektórych pacjentów może się
jednak ujawnić zmniejszona odpowiedź F na 72-godzinną stymulację ACTH [23]. Mechanizm prawidłowego poziomu F nie doczekał się jeszcze wyjaśnienia, bo nie może to być spowodowane tylko
faktem, że jest zachowana częściowo aktywność
21-hydroksylazy.
U wszystkich badanych pacjentek z nk-wpn potwierdzono prawidłowe dobowe wydalanie metabolitów F, a u pacjentek w okresie dojrzewania nawet
lekko podwyższone względem grupy kontrolnej (tab.
II). Potwierdzono istotną (p<0.001) dodatnią korelację, dobowego wydalania metabolitów F z dobowym
wydalaniem metabolitów 17-OHP, 21-DF oraz androstendionu (ryc.1, 2, 3). Wynika z tego, że nadnercza osób z nk-wpn odpowiadają zwiększonym wytwarzaniem F na stymulację przez ACTH, kompensując deficyt enzymatyczny na drodze jego syntezy.
Huerta i wsp. potwierdzili, że większość pacjentów z
nk-wpn wykazuje wzmożone odpowiedzi Δ4 i Δ5-steroidów i 11-dezoksykortyzolu (prekursor F) na stymulację przez ACTH. U badanych przez nich pacjentów z nk-wpn, podstawowe poziomy F w surowicy były prawidłowe lub nawet podwyższone, a odpowiedź F po ACTH nie różniła się od grupy kontrolnej. Autorzy postawili hipotezę, że jak się wydaje, wzmożona wrażliwość nadnerczy na ACTH,
może stanowić odbicie zwiększonej masy nadnerczy u pacjentów z nk-wpn [24]. Potwierdzeniem
tej hipotezy jest fakt, że u około 40% pacjentów z
Ryc. 2. Zależność między dobowym wydalaniem metabolitów
F (THE+THF+allo-THF) i 17-OHP (PT+17-OHPN) u kobiet
z nk-wpn, z pokazaniem linii regresji. Osie x i y są w skali log
Fig. 2. Scater plot of daily excretion of F metabolites
(THE+THF+allo-THF) against daily excretion of 17-OHP
metabolites (PT+17-OHPN) in NC-CAH women. The linear
regression line is drawn
Ryc. 3. Zależność między dobowym wydalaniem metabolitów
F (THE+THF+allo-THF) i 21-DF (PNT) u kobiet z nk-wpn,
z pokazaniem linii regresji. Osie x i y są w skali log
Fig. 3. Scater plot of daily excretion of F metabolites
(THE+THF+allo-THF) against daily excretion of 21-DF
metabolites (PNT) in NC-CAH women. The linear regression
line is drawn
nk-wpn potwierdza się przerost nadnerczy lub gruczolaki [9,10]. Inne badania potwierdziły, że wytwarzanie u pacjentów z nk-wpn progestagenów i androgenów jest częściowo niezależne od stymulacji przez
ACTH i oprócz łagodnego przerostu kory nadnerczy,
należy też wziąć pod uwagę nietypową kinetykę enzymów. Wynika z tego, że poziom 17-OHP może nie
być dobrym wskaźnikiem skuteczności leczenia substytucyjnego [25].
Dehydrogenaza 11β-hydroksysteroidowa typu
1(11β-HSD1) jest enzymem, działającym jako
15
Praca oryginalna
Ryc. 4. Zależność między dobowym wydalaniem metabolitów
F (THE+THF+allo-THF) i androstendionu (11-hydroksyandrosteron) u kobiet z nk-wpn, z pokazaniem linii regresji
Fig. 4. Scater plot of daily excretion of F metabolites
(THE+THF+allo-THF) against daily excretion of
androstenedione metabolite (11-hydroxyandrosterone)
in NC-CAH women. The linear regression line is drawn
okso-reduktaza, przekształcającym nieaktywny
kortyzon do F, kontrolując jego ilość, która zwiąże się z receptorem glikokortykoidowym. Obniżenie jej aktywności zaburza metabolizm i zwiększa
klirens F, powodując hipokortyzolemię i tym samym podwyższenie wydzielania ACTH [26]. U pacjentów z nk-wpn obniżona aktywność 11β-HSD1
może być dodatkowym czynnikiem, stymulującym
poprzez ACTH zwiększone wytwarzanie androgenów. Potwierdzenie u 31.4 % pacjentów z nk-wpn
obniżonej aktywności 11β-HSD1 (tab. III) zwraca uwagę na ten dodatkowy czynnik, który nie pozwala efektywnie wykorzystać F. Leczenie substytucyjne hydrokortyzonem tych pacjentów może być
mniej efektywne. Na możliwy wpływ obniżonej aktywności 11β-HSD1 na brak prawidłowych efektów
leczenia hydrokortyzonem zwrócono uwagę u pacjentów z klasyczną postacią wpn w okresie dojrzewania [27].
W analizowanej grupie 52 pacjentek z nk-wpn,
dobowe wydalanie metabolitu aldosteronu THAldo
u części z nich, tzn. tylko młodszych pacjentek w
wieku 2.5-17 lat, było istotnie wyższe w porównaniu do grup kontrolnych zdrowych dzieci, potwierdzając zwiększoną syntezę aldosteronu. Sugeruje to
istnienie mechanizmu kompensowania epizodów
subtelnego niedoboru aldosteronu (lekki niedobór
21-hydroksylazy także na drodze syntezy mineralokortykoidów) w okresie 24 godzin, poprzez pobudzanie osi renina-aldosteron.
16
Endokrynol. Ped., 7/2008;1(22):7-16
Bardzo mało jest badań na temat funkcjonowania osi renina-aldosteron u pacjentów z nk-wpn.
Prawidłowe stężenie aldosteronu w osoczu i brak
utraty soli u tych pacjentów był dotąd jedynym
dowodem na prawidłową syntezę mineralokortykoidów [5]. Fiet i wsp. po raz pierwszy w 1989 r.
zasugerowali ma podstawie wyników swoich badań, że u pacjentów z nk-wpn istnieje dyskretny
niedobór 21-hydroksylazy także na drodze syntezy mineralokortykoidów [28]. Dopiero w 2007 r.
przeprowadzono kompleksowe badania oceniające utratę soli na bardzo dużej liczebnie grupie pacjentów z różnymi postaciami wpn. Jako wskaźnik
utraty soli przyjęto stosunek stężenia aldosteronu
do aktywności reninowej osocza. Wykazano nieprawidłowe wartości w porównaniu do grupy kontrolnej we wszystkich postaciach wpn, nie tylko z
utratą soli, ale także z prostą wirylizacją i także
nk-wpn. Niedobór aldosteronu występuje, chociaż
o różnym stopniu nasilenia, we wszystkich postaciach wpn. Pacjenci z łagodnym defektem 21-hydroksylazy potrzebują zwiększonej aktywności reninowej osocza, aby stymulować lekko zaburzoną
syntezę aldosteronu, zabezpieczającą prawidłowe
stężenie sodu. Ponieważ nie ma do chwili obecnej
doniesień o utracie soli u pacjentów z nk-wpn, autorzy stwierdzają, że nie ma potrzeby podawania
im mineralokortykoidów [29].
Potwierdzenie nk-wpn u starszej, asymptomatycznej kobiety z gruczolakami nadnerczy, wykrytymi jako incidentaloma, potwierdza konieczność wykluczenia takiej etiologii w diagnostyce różnicowej
nawet u starszych kobiet [12]. U pacjentów z wpn
obserwuje się regresję zmian w nadnerczach pod
wpływem leczenia glikokortykoidami [10, 12, 30].
Wniosek
W diagnostyce różnicowej objawów androgenizacji u płci żeńskiej, niezalęznie od wieku pacjentek,
jak również w przypadkach incidentaloma w nadnerczach, należy zawsze brać pod uwagę nk-wpn. Obserwowana u pacjentek z nk-wpn progresja objawów
klinicznych wraz z wiekiem, pochodzących wcześniej z dysfunkcji nadnerczy, prowadzi z czasem u
wielu pacjentek do zaburzenia czynności gonad.
Dobowe wydalanie metabolitów kortyzolu jest
prawidłowe u pacjentek z nk-wpn. Ponieważ koreluje ono dodatnio, istotnie z wydalaniem metabolitów 17-OHP, 21-DF i androstendionu sugeruje to, że nadnercza pacjentek z nk-wpn odpowiadają zwiększonym wytwarzaniem F na stymulację
Ewa Maria Maunowicy – Nieklasyczna postać wrodzonego przerostu kory nadnerczyz powodu niedoboru 21-hydroksylazy (nk-wpn) u dziewczynek...
ACTH, kompensując deficyt enzymatyczny na drodze jego syntezy.
Zwiększone istotnie dobowe wydalanie metabolitu aldosteronu THAldo u pacjentek w wieku
2.5-17 lat w porównaniu do grup kontrolnych zdro-
wych dziewczynek, wskazuje na istnienie mechanizmu kompensacji dyskretnego niedoboru 21-hydroksylazy, występującego także na drodze syntezy
mineralokortykoidów.
PIŚMIENNICTWO/REFERENCES
[1]
[2]
[3]
[4]
[5]
[6]
[7]
[8]
[9]
[10]
[11]
[12]
[13]
[14]
[15]
[16]
[17]
[18]
[19]
[20]
[21]
[22]
[23]
Moran C., Knochenhauer E. S., Azziz R.: Non-classic adrenal hyperplasia in hyperandrogenism: a reappraisal. J. Clin. Endocrinol. Invest., 1998: 21, 707-720.
Azziz R., Hincapie L. A., Knochenhauer E. S. et al.: Screening for 21-hydroxylase-deficient nonclassic adrenal hyperplasia
among hyperandrogenic women: a prospective study. Fertil Steril. 1999: 72, 915-925.
Azziz R., Knochenhauer E. S., Moran C.: Androgen excess in women: experience with over 1000 consecutive patients.
J. Clin. Endocrinol. Metab., 2004: 89, 453-62.
Carmina E., Rosato A., Janni’ A. et al.: Relative prevalence of different androgen excess disorders in 950 women referred
because of clinical hyperandrogenism. J. Clin. Endocrinol. Metab., 2006: 9, 2-6.
New M. I.: Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 2006: 91,
4205-4214.
Małunowicz E., Ginalska-Malinowska M., Szypulska M., Romer T. E.: Nonclassic congenital adrenal hyperplasia due to
21-hydroxylase deficiency: frequency in children with precocious pubarche and in adolescent girls with menstrual disturbances and/or hirsutism. Ann. Diagn. Paediatr. Pathol., 1999: 2, 1-7.
Moran C., Azziz R., Carmina E. et al.: 21-hydroxylase-deficient nonclassic adrenal hyperplasia is a progressive disorder:
A multicenter study. Am. J Obstet. Gynecol., 2000: 183, 1468-1474.
Levine L. S., Dupont B., Lorenzen F. et al.: Cryptic 21-hydroxylase deficiency in families of patients with classical congenital
adrenal hyperplasia. J Clin. Endocrinol. Metab., 1980: 51, 1616-1324.
Azziz R., Kenney P. H.: Magnetic resonance imaging of the adrenal gland in women with late-onset adrenal hyperplasia.
Fertil. Steril., 1991: 56, 142-144.
Jaresh S., Kornely E., Kley H. K. et al.: Adrenal incidentaloma and patients with homozygous or heterozygous congenital
adrenal hyperplasia. J. Clin. Endocrinol. Metab. 1992: 74, 685-689.
Kohn B., Levine L. S., Pollack M. S. et al.: Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital
adrenal hyperplasia. J. Clin. Endocrinol. Metab., 1982: 55, 817-827.
Falhammar H., Thorèn M.: An 88-year-old woman diagnosed with adrenal tumor and congenital adrenal hyperplasia: connection or coincidence. J. Endocrinol. Invest., 2005: 28, 449-453.
Wilson R. C., Mercado A. B., Cheng K. C. et al.: Steroid 21-hydroxylase deficiency: genotype may not predict phenotype.
J. Clin. Endocrinol. Metab., 1995:80, 2322-2329.
Azziz R.: 21-hydroxylase deficient nonclassic adrenal hyperplasia. Endocrinologist 1995: 5, 297-303.
Azziz R., Hincapie L. H., Knochenhauer E. S. et al.: Screening for 21-hydroxylase-deficient nonclassic adrenal hyperplasia
among hyperandrogenic women: a prospective study. Fertil. Steril., 1999:72, 915-925.
Dewailly D.: Nonclassic 21-hydroxylase deficiency. Semin. Reprod. Med., 2002: 3, 243-248.
Homoki J., Solylom J., Teller W. M.: Detection of late onset steroid 21-hydroxylase deficiency by capillary gas chromatographic profiling of urinary steroids in children and adolescents. Eur. J. Pediatr., 1988:.147, 257-263.
Török D., Halász Z., Garami M.: Limited value of serum steroid measurements in identification of mild form of 21-hydroxylase deficiency. Exp. Clin. Endocrinol. Diabetes. 2003: 111, 27-32.
Shackleton C. H. L.: Mass spectrometry in the diagnosis of steroid-related disorders and in hypertension research. J. Steroid Biochem. Molec. Biol., 1993: 45, 127-140.
Feuillan P., Pang S., Schürmeyer T. et al.: The hypothalamic-pituitary-adrenal axis in partial (late-onset) 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab., 1988: 67, 154-160.
Carmina E., Lobo R.: Pituitary-adrenal responses to corticotropin -releasing factor in late onset 21-hydroxylase deficiency.
Fertil. Steril., 1990: 54, 79-83.
Moreira A. C., Elias L. L. K.: Pituitary-adrenal responses to corticotropin –releasing hormone in different degrees of adrenal
21-hydroxylase deficiency. J. Clin. Endocrinol. Metab., 1992: 74, 198-203.
Kater C., Biglieri E. G., Wajchenberg B.: Effects of continued adrenal corticotropin stimulation on the mineralocorticoid
hormones in classical and nonclassical symbol virilizing types of 21-hydroxylase deficiency. J Clin. Endocrinol. Metab., 1985:
60, 1057-62.
17
Praca oryginalna
Endokrynol. Ped., 7/2008;1(22):9-18
[24] Huerta R., Devailly D., Decanter C. et al.: Adrenocortical hypersensivity to adrenocorticotropin hormone: a mechanism
favoring the normal production of cortisol in 21-hydroxylase-deficient nonclassic adrenal hyperplasia. Fertil. Steril., 2000: 74,
329-334.
[25] Sánchez L. A., Morán C., Reyna R. et al.: Adrenal progestogen and androgen production in 21-hydroxylase-deficient
non-classic adrenal hyperplasia is partially independent of adrenocorticotropin stimulation. Fertil. Steril. 2002: 77, 750-753.
[26] Draper N., Stewart P. M.: 11β-hydroxysteroid dehydrogenase and pre-receptor regulation of corticosteroids hormone action.
J. Endocrinol 2005: 186, 251-271.
[27] Charmandari E., Hindmarsh P. C., Johnston A. et al.: Congenital adrenal hyperplasia due to 21-hydroxylase deficiency:
Alterations in cortisol pharmacokinetics at puberty. J. Clin. Endocrinol. Metab. 2001: 86, 2701-2708.
[28] Fiet J., Gueux B., Raux-Femay M-C.: Increased plasma 21-deoxycorticosterone (21-DB) levels in late-onset adrenal 21-hydroxylase deficiency suggest a mild defect of the mineralocorticoid pathway. J. Clin. Endocrinol. Metab. 1989: 68, 542-547.
[29] Nimkarn S., Lin-SU K., Berglind N. et al.: Aldosterone-to-renin ratio as a marker for disease severity in 21-hydroxylase
deficiency congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab., 2007: 92, 137-142.
[30] Giacaglia L. R., Mendonca B. B., Madureira G. et al.: Adrenal nodules in patients with congenital adrenal hyperplasia due to
21-hydroxylase deficiency: regression after adequate hormonal control. J. Pediatr. Endocrinol. Metab., 2001: 14, 415-419.
18