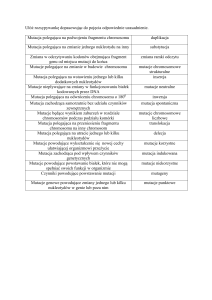
MONGOLIZM (zespół Downa),
choroba uwarunkowana genetycznie, której istotą jest dodatkowy chromosom 21; wrodzony niedorozwój umysłowy
różnego stopnia, połączony z charakterystycznymi zmianami fiz., jak: mała, spłaszczona czaszka, skośne szpary
powiekowe, szeroko rozstawione oczy, wiotkie stawy i in.
Trisomia przy 21 parze chromosomów jest powodem wystąpienia mongolizmu, czyli zespołu Downa. Częstość
mutacji tej wzrasta wraz z wiekiem matki. Dzieci z zespołem Downa wykazują niski wzrost, szerokie, skośne
rozstawienie oczu z fałdą nakątną, spłaszczony i mały nos, suchość skóry, upośledzenie umysłowe i ruchowe,
płaskość twarzy, niskie osadzenie małżowin usznych, krótkie i zakrzywione palce, wady wsierdzia.
Trisomia przy 13 parze chromosomów – zespół Pataua – mutacja przejawia się rozszczepem wargi i podniebienia,
degeneracją małżowin usznych, zaciśniętymi pięściami, degeneracją mózgowia i uwstecznieniem oczu
(mikroftalmia), wnętrostwem i wadami wrodzonymi serca. Połowa dzieci umiera w I miesiącu po urodzeniu.
Trisomia przy 18 parze chromosomów – zespół Edwardsa – mutacja jest z reguły letalna, większość (95%)
przypadków kończy się śmiercią w łonie matki (poronienie). Urodzone osobniki są upośledzone umysłowo, mają
małą, cofniętą brodę, wydatną potylicę, łyżwiaste stopy, nisko osadzone uszy, zaciśnięte pięści, wady rozwojowe
nerek i serca.
47, XYY, czyli dodatkowy chromosom Y – mutacja przejawia się wysokim wzrostem, agresywnością i frustracją
osobnika. Obniżenie inteligencji jest rzadkością.
1
47, XXY, czyli dodatkowy chromosom X – zespół Klinefeltera - mutacja przejawia się bezpłodnością, małymi
genitaliami, ginekomastią, silnie wydłużonymi kończynami, obniżeniem inteligencji, skoliozą i osteoporozą.
47, XXX, czyli dodatkowy chromosom X u kobiet – mutacja przejawia się obniżona inteligencją.
Monosomia 45, X – brak jednego chromosomu płciowego - zespół Turnera – mutacja manifestuje się
niedrożnością naczyń chłonnych, niskim wzrostem, brakiem menstruacji, szeroką klatką piersiową, dużym
rozstawem piersi, krótką płetwiastą szyją ze sporymi fałdami skóry, zwłóknieniem jajników, zwężeniem aorty,
wadami rozwojowymi nerek i serca.
Zwielokrotnienie liczby chromosomów w obrębie tego samego gatunku prowadzi do powstania form
autoploidalnych.
Mukowiscydoza [Cystic Fibrosis] jest chorobą genetycznie uwarunkowaną przekazaną przez rodziców. Ponieważ
jest to schorzenie genetyczne chory musi urodzić się z nią. Obecnie nie istnieje lek przeciw Mukowiscydozie, ale to
może się zmienić. Lepsze zrozumienie przyczyn choroby oraz szybki rozwój metod, które pozwolą na ich usunięcie,
zwiększają szansę wyleczenia. Zanim to nastąpi, coraz skuteczniej leczone są objawy, co pozwala zwolnić postęp
choroby.
Mukowiscydoza jest chorobą częstą. W wielu krajach przeciętnie jedno na 2.500 rodzących się dzieci choruje na
Mukowiscydozę. Przykładowo, jeżeli w kraju rodziłoby się 10.000 dzieci rocznie, czworo z nich byłoby chore na
Mukowiscydozę.
Dysplazja Ektodermalna. Każdy związek wad pochodnych ektodermy przedstawia inny typ ED. Do tej pory
zidentyfikowano co najmniej 150 różnych form, w zależności od zasięgu i surowości symptomów.
U dzieci dotkniętych syndromem ED mogą występować następujące objawy:
nieobecne lub upośledzone gruczoły potowe
anomalie dentystyczne (brak zawiązków zębowych, brakujące zęby, upośledzony rozwój szkliwa
zębowego lub jego zupełny brak, niedorozwiniecie kształtu zębów, odbarwienia itp.)
źle ukształtowane lub brakujące palce u rak i nóg (EKTRODAKTYLIA- małe palce, palce nadliczbowe,
źle ukształtowane, rozszczep dłoni lub stóp-CLEFT )
źle ukształtowane lub nieobecne paznokcie u rąk i nóg
źle funkcjonujące błony śluzowe
rozszczep wargi i podniebienia
wady słuchu
wady wzroku (np. nadwrażliwość na światło słoneczne - fotofobia)
wady kanalików łzowych (brak łez)
wady owłosienia (włosy na głowie rzadkie i świetliste, brak pigmentu włosa, anomalie w budowie, brwi,
rzęsy i inne włosy na ciele mogą być nieobecne lub rzadkie)
ZABURZENIA GENETYCZNE PRZED 25 ROKIEM ŻYCIA
Genetycy potrafią określić już na etapie rozwoju embrionalnego narażenie na schorzenia, jakie ujawniają się dopiero
po kilkudziesięciu latach od przyjścia na świat. Przykładem są pląsawica Huntingtona, zaburzenia hemoglobiny,
miopatia Steinera, a nawet niektóre rodzaje nowotworów – rak piersi, jelita grubego, nerki oraz siatkówki oka.
Od niedawna w Instytucie Matki i Dziecka w Warszawie wykonywane są też badania wykrywające u dzieci
rdzeniowy zanik mięśni (SMA) – druga po mukowiscydozie najczęściej występującą chorobą genetyczną. Z badań
tych powinniśmy skorzystać przede wszystkim te osoby, w których rodzinie stwierdzono już taką wadę. A jest ich
dość dużo. Genetycy oceniają, że 2 – 3 % dzieci przychodzi na świat z jakimś zaburzeniem rozwojowym, które aż w
85 % jest uwarunkowane genetycznie. Poważna choroba dziedziczna ujawnia się, u co dwudziestej osoby przed 25
rokiem życia. Tego zagrożenia nie warto ignorować, tym bardziej, że tylko wczesne wykrycie schorzenia umożliwia
skuteczne leczenie i rehabilitację.
Tak jest w przypadku rdzeniowego zaniku mięśniowego, występującego u jednego dziecka na 15 – 20 tys. urodzeń.
W ciężkiej postaci choroba powoduje zgon tuż po narodzinach – w ciągu 3 – 4 lat. Bardziej skuteczne jest leczenie
łagodnej jej odmiany, powodującej niepełnosprawność ruchową dużego stopnia, przy intelekcie wyższym niż
przeciętny. Dzieciom chorym na SMA, ze względu na ich wiotkość ciała, szczególnie grożą skrzywienia kręgosłupa
2
i różnego rodzaju przykurcze.
Niestety, nie można jeszcze wykrywać nosicieli SMA – ludzi, którzy nie chorują, ale przenoszą na potomstwo
wadliwy gen (znajdujący się w chromosomie 5) wywołujący u nich chorobę. W Polsce, co 40 osoba jest jej
nosicielem, a ryzyko ponownego wystąpienia SMA w rodzinie dotkniętej zaburzeniem sięga aż 25 % - pierwszym
przypadkom chorób genetycznych w rodzinie na razie nie można zapobiec, ale u następnego dziecka można
przynajmniej wcześnie stwierdzić lub ją wykluczyć. Genetycy zalecają, zatem, by ich członkowie poddali się testom
genetycznym.
Podobnie można już wcześnie wykrywać skłonności genetyczne do chorób nowotworowych: raka jajników, piersi,
jelita grubego. Trwają też zaawansowane badania nad genetyczną diagnostyką raka prostaty, nerki, mózgu,
gruczołów wydzielania wewnętrznego, trzustki, żołądka, płuc i tarczycy. W Polsce powstaje cała sieć genetycznych
poradni onkologicznych, która obejmie 16 ośrodków, m.in. w Bydgoszczy, Gdańsku, Gliwicach, Krakowie, Łodzi,
Poznaniu, Warszawie i Wrocławiu. Ocenia się, bowiem, że dziedziczne predyspozycje jedynie do raka piersi i jelita
grubego wykazuje 0,5 – 1 mln Polaków.
3