Child Neurology
Neurologia
Dziecięca
ARTYKUŁ REDAKCYJNY/EDITORIAL
Vol. 15/2006 Nr 29
Znaczenie badań farmakokinetycznych
leków przeciwpadaczkowych w leczeniu
padaczki u dzieci i młodzieży
The importance of pharmacokinetic studies
of antiepileptic drugs in the treatment of epilepsy
in children and adolescents
Barbara Steinborn
Katedra i Klinika Neurologii Wieku Rozwojowego Akademii Medycznej im. Karola Marcinkowskiego w Poznaniu
Kierownik: dr hab. med. Barbara Steinborn
Streszczenie
Słowa kluczowe: leki
przeciwpadaczkowe
– parametry farmakokinetyczne
W pracy przedstawiono korzyści wynikające ze znajomości farmakokinetyki leków przeciwpadaczkowych (LPP). Poznanie terapeutycznych stężeń, okresu biologicznego półtrwania (t1/2), klirensu (Cl), oraz zmian wiązania się z białkami surowicy krwi są w praktyce klinicznej niezwykle
użyteczne. Rozpoczęcie i kontynuacja leczenia lekami przeciwpadaczkowymi (LPP) są znacznie
ułatwione dzięki znajomości farmakokinetyki i możliwych interakcji. Podano także wartości oraz
porównanie tych parametrów dla leków konwencjonalnych i nowszych.
Abstract
Key words: Antiepileptic
drugs – pharmacokinetic
parameters
Usefulness of knowledge of antiepileptic drugs (AEDs) pharmacokinetics were presented in this
paper. The knowledge of therapeutic concentration range, half-life time (t1/2), clearance rate (Cl),
and changes in binding with the proteins of blood serum are significantly useful in clinical practice.
Their introduction and continuation of AEDs treatment are significantly facilitated if wide knowledge
of pharmacokinetics and possible interactions is administered and applied. The pharmacokinetic
parameters of conventional and newer AEDs were presented and compared as well.
Korzyści ze znajomości
farmakokinetyki
Celem leczenia padaczki jest tłumienie napadów padaczkowych i jednoczesne unikanie występowania objawów niepożądanych związanych ze stosowaniem leków
przeciwpadaczkowych (LPP). Osiągniecie tego jest możliwe dzięki odpowiedniemu rozpoznaniu napadów padaczkowych/zespołów padaczkowych i odpowiedniemu
doborowi LPP. Poza tym ważne jest precyzyjne określenie dawki (u dzieci dawki leków ustala się w oparciu o
masę ciała) i sposobu wprowadzenia leku oraz utrzymanie odpowiedniego stężenia LPP w surowicy krwi [1].
Kontrola napadów u poszczególnych chorych leczonych takimi samymi lekami i przy osiągnięciu podobnych stężeń w surowicy krwi może być niejednakowa. Wynika to ze zróżnicowanej osobniczej reakcji na
leczenie. Czynnikami, które wpływają na to, są: zróżnicowanie genetyczne, dieta, współwystępowanie innych
chorób, dodatkowe leczenie, a także wiek i płeć [2–5].
Vol. 15/2006, Nr 29
Rozpoczęcie i kontynuacja leczenia LPP są ułatwione dzięki znajomości farmakokinetyki i interakcji
lekowych. Selektywne pomiary stężeń leków w płynach biologicznych, zwłaszcza przy terapii kombinowanej, mogą poprawić postępowanie lecznicze [6].
Przy wyborze LPP najczęściej bierze się pod uwagę
jego skuteczność i bezpieczeństwo. Te dwie cechy są
możliwe do oceny po osiągnięciu stężenia stacjonarnego. Przy tej samej skuteczności i podobnych objawach
niepożądanych wybiera się lek o lepszym profilu farmakokinetycznym i łatwiejszej formie użycia [7]. W
klinice najłatwiej byłoby podawać lek o tzw. idealnym
profilu farmakokinetycznymi, który charakteryzuje się
przede wszystkim liniową kinetyką [8]. W praktyce
oznacza to łatwą przewidywalność zmian stężeń leku
w zależności od zmian dawek. Te dwie wartości są
wprost proporcjonalne. Idealny lek cechuje się także
wysoką, całkowitą biodostępnością i szybką absorpcją.
Poza tym nie powinien wiązać się z białkami surowicy
krwi lub wiązanie to powinno zachodzić w jak najniż
Barbara Steinborn
szym stopniu. Objętość dystrybucji (Vd) powinna być
wartością stałą w całej populacji, co pozwalałoby na
obliczenie dawki nasycającej, tzw. loadning dose. Tak
samo stała powinna być jego eliminacja, niezależna
od czasu i dawek. Nerkowy klirens (Cl) powinien korelować z Cl kreatyniny i być w całej populacji także
względnie stałą wartością. Lek taki nie powinien być
metabolizowany w wątrobie i nie powinien mieć także aktywnych metabolitów. Jeśli natomiast metabolit
jest aktywny, to prekursor leku nie powinien wykazywać aktywności przeciwdrgawkowej. Idealny lek nie
wchodzi w interakcje lekowe [1]. Wśród nowych LPP
niektóre mają profil farmakokinetyczny zbliżony do
idealnego. Według Patsalosa takim preparatem może
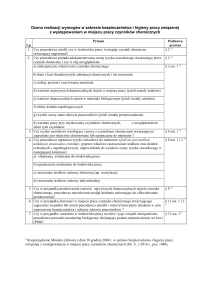
być levetiracetam (LEV) [8]. Na podstawie skali punktowej zaproponowanej przez tego autora LEV i VGB
spełniają takie kryteria w 94% (tab. I). Bardzo dobrze
wchłaniają się z przewodu pokarmowego, można je podawać raz lub dwa razy na dobę i mają liniową kinetykę. VGB nie ulega metabolizmowi, a LEV w nieznacznym stopniu. Z kolei LEV nie wchodzi w interakcje, a
VGB w niewielkim stopniu [8]. Pozostałe, a zwłaszcza
tzw. konwencjonalne LPP, mają kinetykę daleką od
idealnej (tab. II, III i IV).
Najbardziej pożądanymi cechami LPP, które ułatwiają ich dawkowanie z punktu widzenia klinicysty,
są przede wszystkim: mała ilość lub brak interakcji,
możliwość stosowania leków 1-2x/dobę oraz dostępność postaci do podań pozajelitowych [7].
Poznanie zakresów stężeń terapeutycznych (C) ma
znaczenie dla oceny efektów klinicznych i objawów
niepożądanych danego leku. Stężenia subterapeutyczne potencjalnie mogą nie zapewnić kontroli napadów.
Z kolei stężenia potencjalnie toksyczne mogą być
przyczyną występowania objawów niepożądanych lub
toksycznych. Zakres terapeutyczny jest wartością statystyczną i dotyczy większości chorych (tab. IV i V).
Efekt kliniczny może wystąpić także przy stężeniach
poniżej normy. Podobnie, jak i objawy niepożądane
mogą pojawić się nawet w zakresach stężeń terapeutycznych [6, 9].
Biologiczny okres półtrwania (t½) daje informacje
o rozmiarach wahań stężeń leku pomiędzy dawkami i
jednocześnie określa czas oraz przedziały dawkowania leku. Oprócz tego, określa także czas potrzebny do
osiągnięcia stężenia stacjonarnego i do zniknięcia leku
z surowicy krwi po jego odstawieniu [5]. W zależności
od częstości podawania LPP zmienia się też stosowanie się pacjentów do przestrzegania zaleceń lekarskich.
Im mniejsza częstotliwość podawania leków w ciągu
doby, tym większe prawdopodobieństwo stosowania
się do zaleceń. LPP powinny być stosowane z częstością, która odpowiada t½, jeśli zaś są to wartości większe
od 24 h to wystarczy stosowanie 1x/dobę. Taki sposób
podawania leków jest kompromisem polegającym na
maksymalizacji efektu i minimalizacji objawów niepożądanych, związanych z występowaniem maksymalnych stężeń LPP [5]. Przy ustalaniu częstości podawania leku należy uwzględnić możliwość zmian t½ w
zależności od wieku pacjenta i stosowania innych leków. Wiek , który determinuje szybkość metabolizmu
leku, a także wydolność wątroby i nerek, powoduje, że
u dzieci t½ są wartościami mniejszymi niż u osób dorosłych.
Oprócz t½, jako parametru farmakokinetycznego
wprowadzono także pojęcie czasu biologicznej aktywności leku czy okresu półtrwania farmakodynamicznego [5]. Zależy on zarówno od okresu półtrwania, jak i
czasu wiązania się z aktywnym receptorem. Dla leków
konwencjonalnych te dwie wartości są praktycznie takie same. Natomiast dla nowych LPP okres aktywności
biologicznej jest dłuższy od t½. Klasyczne parametry
farmakokinetyczne mają mniejszą wartość dla oceny właściwości biologicznych leków [5]. Ta różnica
związana jest ze specyficznymi mechanizmami działania nowych LPP. Cechy takie wykazuje VGB, która
wiąże się kowalentnie z transaminazą GABA (GABAT) unieczynniając ten enzym, co prowadzi do wzrostu
stężenia GABA. Czas działania biologicznego VGB
wynosi kilka dni, a t½ ok. 5 h. Pojawienie się ponowne
GABA-T związane jest z jej resyntezą, a nie dysocjacją
z VGB [5]. W przypadku VPA, którego t½ wynosi 6-9
h, obserwowano, że efekt związany z tłumieniem wyładowań czynności bioelektrycznej mózgu pod wpływem
fotostymulacji pojawia się dopiero po 5 dniach [10].
Takie zróżnicowanie okresu biologicznej aktywności
leku i t½ ma swoje konsekwencje kliniczne, zwłaszcza
w podjęciu decyzji co do czasu podawania takich leków, jak: VGB, TGB czy VPA, gdzie te dwie wartości
nie są równe. W takich sytuacjach czas dawkowania
nie musi odpowiadać t½ [7].
Znajomość parametrów farmakokinetycznych stosowanych leków pozwala także na obliczenie dawki
leku, która zapewni szybkie nasycenie. Ma to miejsce
w sytuacjach wymagających szybkiego osiągnięcia stanu stacjonarnego. Znajomość wartości objętości dystrybucji (Vd) i wymaganego stężenia terapeutycznego
są podstawą do obliczenia tzw. dawki nasycającej (LD)
[5]. Dawka potrzebna do osiągnięcia takiego stanu wyrażona jest iloczynem:
Vd [l/kg] x C[mg/l] = LD [mg/kg].
W takiej sytuacji może pojawić się także problem
występowania objawów niepożądanych związanych z
szybkim nasycaniem. Dla większości LPP proponuje
się powolne ich wprowadzanie, tj. stopniowe zwiększanie dawek, aż do dawki terapeutycznej, tak aby zmniejszyć ryzyko wystąpienia objawów niepożądanych.
W taki sposób postępuje się przy wprowadzaniu do leNeurologia Dziecięca
Znaczenie badań farmakokinetycznych leków przeciwpadaczkowych w leczeniu padaczki u dzieci i młodzieży
Tab. I. Porównanie właściwości farmakologicznych LPP. The comparison of pharamcologic properties of AEDs.
[8]
LPP
Wchłanianie
po podaniu doustnym
Dawkowanie
Kinetyka
Metabolizm
LEV
VGB
GBP
TPM
LTG
TGB
3
3
3
2
3
2
3
3
1-2
3
3
2
3
3
2
3
2
3
2
3
3
3
1
1
Interakcje lekowe
i
3
3
3
1
1
2
% leku idealnego*
ii
3
2
3
1
2
1
94
94
86
72
67
61
* Wyliczony poprzez dodanie do siebie wszystkich uzyskanych punktów za każdą cechę (maksymalnie 3 pkt.),
podzielonych następnie przez maksymalną liczbę 18.
Tab. II. Charakterystyka LPP (konwencjonalnych i nowych) pod względem łatwości użycia.The attributes of AEDs
which improved of management in therapy. [33]
DawkoLiniowa
wanie 1-2
kinetyka
x na dobę
Leki
konwencjonalne
CBZ
VPA
PHT
PB
ESM
BZD
Nowe
leki
FBM
GBP
LTG
OCBZ
TGB
TPM
VGB
ZNS
LEV
Brak znaczącego
wiązania
z białkami
(<80%)
Brak znaczącego
metabolizmu
Nerkowe
drogi eliminacji
Brak interakcji
Brak potrzeby
stopniowego zwiększania dawek do
najniższej efektywnej dawki
+1
+1
+
+
+
+
+
+
+
+
+
+
-
-
-
-
+
+
-
+
+
+
-2
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+3
+
+
+
+
+
+
Tylko dla preparatów o przedłużonym działaniu.
Oficjalne zalecenia 3x/dobę, ale w badaniach klinicznych potwierdzono dobre efekty w dawkach 2x/dobę.
3
Niestałe zmniejszenie poziomu PHT o 15-30%.
1
2
Vol. 15/2006, Nr 29
Barbara Steinborn
Tab. III. Porównanie parametrów farmakokinetycznych nowych LPP. The comparison of pharmacokinetic parametres of new AEDs.
LPP
Wiązanie
z białkami (%)
T1/2 (h)
Miejsce eliminacji
Uwagi kliniczne
GBP
0
4-6
nerki – 100%
wykazuje zależną od dawki absorpcję
LTG
55
15-30
wątroba – 90%
klirens (drogą wiązania z kwasem glukuronowym)
zwiększają LPP indukujące enzymy, zmniejsza zaś
VPA; metabolity są nieaktywne
TPM
9-17
15-23
nerki – 40-70%
LEV
lek częściowo metabolizowany w wątrobie; metabolizm wątrobowy nasilają LPP indukujące enzymy;
metabolity są nieaktywne
0
6-8
OCBZ
40
4-9
nerki – 66%
hydroliza grupy acetamidowej –34%
wątroba – 70%
TGB
96
4-7
wątroba – 98%
metabolizm odbywa się poza wątrobą; metabolity są
nieaktywne
metabolizm przebiega z wytworzeniem 10-hydroksykarbazepiny (MHD), która jest głównym aktywnym
metabolitem
utlenienie do nieaktywnych metabolitów
ZNS
40-60
24-60
wątroba – 70%
klirens zwiększają LPP indukujące enzymy
Tab. IV. Parametry farmakokinetyczne LPP (konwencjonalnych). Pharmacokinetic parametres of conventional
AEDs. [7]
Lek
F%
Karbamazepina
Klobazam
Klonazepam
Ethosuximid
Fenobarbital
Fenytoina
Primidon
Kwas walproinowy
75-85
>90
>90
>90
>90
>90
>90
>90
Tmax (h) Vd (L/kg)
4-12
1-4
1-4
1-4
0,5-4
2-12
2-4
1-8¶
Wiązanie
z białkami
0,8-2
75
3,0
0,65
0,55
0,75
0,75
0,16
85
<10
45
90
<20
70-93║
T1/2 (h)
Tss d
Stężenie
terapeutyczne
mg/L
μmol/L
20-50†
10-30
20-40
30-60
65-110
10-60║
8-15
5-15
20-30†
3-12
12-50
6
7
15-20
15-20
20-75‡
40-100
10-30
3-20
60-250§
300-700
40-130
12-80
2
50-100
350-700
Dawka
(mg/kg/d)
10-30
0,5-2,0
0,1-0,15
10-40
2-5
5-10
10-20
15-30
F – biodostępność, Tmax – czas do osiągnięcia maksymalnego stężenia, Vd – objętość dystrybucji, T1/2 – okres
półtrwania, † – okres półtrwania w stanie stacjonarnym po osiągnięciu autoindukcji, ║– zależność od stężenia, ¶
– absorpcja (dojelitowych) tabletek jest opóźniona, ‡ – μg/L, § – nmol/L.
10
Neurologia Dziecięca
Znaczenie badań farmakokinetycznych leków przeciwpadaczkowych w leczeniu padaczki u dzieci i młodzieży
Tab. V. Parametry farmakokinetyczne LPP (nowych). Pharmacokinetic parameters of new AEDs. [36]
Lek
Gabapentyna
Lamotrygina
Levetiracetam
Oxkarbazepina
10-OH-karbamazepina
Tiagabina
Topiramat
Wigabatryna
Zonisamid
F%
Wiązanie z
Tmax (h) Vd (L/kg)
białkami
35-60
>90
>90
>95
2-3
1-3
1-2
1-2
0,85
1,0
0,6
>90
>80
80
1-2
1-4
0,5-2
2-5
1,4
0,65
0,8
1,5
0
55
<10
15
55
T1/2 (h)
Tss d
5-9
15-60
6-8
2
1-2
3-10
2
96
12-30
5-7
50-70
1-2
3-5
2
10-15
Stężenie terapeutyczne
mg/L
μmol/L
4-16
2-20
20-100
8-80
2-25
6-75
20-30
Dawka
(mg/kg/d)
30-40
1-15
20-60
15-30
0,1-1
5-9
40-100
4-8
F – biodostępność, Tmax – czas do osiągnięcia maksymalnego stężenia, Vd – objętość dystrybucji, T1/2 – okres
półtrwania, † – okres półtrwania w stanie stacjonarnym po osiągnięciu autoindukcji, ║– zależność od stężenia, ¶
– absorpcja (dojelitowych) tabletek jest opóźniona, ‡ – μg/L, § – nmol/L.
czenia: LTG, CBZ, TPM, TGB i ZNS. W przypadku
OXC, PHT, VPA, GBP i LEV, jak podsumowuje Ferrendelli, można od razu zastosować dawkę terapeutyczną lub tzw. nasycającą [11]. Mechanizm tej rozbieżności nie jest wyjaśniony. Fenomen ten, jak interpretuje
sam autor, jest użyteczny w przypadku konieczności
szybkiej kontroli napadów.
Stosowanie LPP odbywa się często w kombinacjach, w postaci bi- lub politerapii. Według danych
przytaczanych prze Rivę i wsp. ok. 70% pacjentów jest
leczonych przy użyciu jednego LPP, u pozostałych istnieje potrzeba stosowania politerapii [2]. Taka sytuacja
prowadzi do wystąpienia interakcji lekowych zarówno farmakokinetycznych, jak i farmakodynamicznych.
Znajomość potencjalnych możliwości i efektów interakcji pomaga klinicystom rozpocząć i kontynuować leczenie, a także ocenić jego skutki [1]. Oprócz potrzeby
poznania t½, Vd, stężeń terapeutycznych, konieczne także jest poznanie dróg eliminacji leków i Cl, dla potrzeb
oceny i interpretacji interakcji [2].
Odmienności farmakokinetyki
leków u dzieci
Zmiany farmakokinetyki LPP należy także spodziewać się w przypadku podawania ich w różnych grupach wiekowych [12, 13]. U dzieci metabolizm leków
jest szybszy niż u dorosłych [12–14].
Poszczególneparametry farmakokinetyczne zmieniają się wraz z dojrzewaniem procesów metabolicznych. Stąd istnieje potrzeba dostosowania zarówno dawek, jak i sposobów podania oraz rodzajów leków, w
zależności od sprawności i dojrzałości poszczególnych
procesów, jakim podlegają leki w ustroju. Zmiany paraVol. 15/2006, Nr 29
metrów farmakokinetycznych zależne od wieku mogą
także być przyczyną większego ryzyka występowania
reakcji idiosynkratycznych u dzieci, zwłaszcza w odniesieniu do VPA i LTG [15]. W przypadku noworodków wchłanianie, dystrybucja oraz wydalanie LPP jest
wolniejsze niż u niemowląt i dzieci starszych [12, 13,
16, 17]. Niewielka powierzchnia wchłaniania, nieregularność ruchów perystaltycznych i niedojrzałość procesów wydzielniczych, zarówno błony śluzowej, jak
i żółci, prowadzi do ograniczenia wchłaniania leków
[12, 13, 16]. Również wchłanianie leków po podaniu
domięśniowym jest gorsze [16]. Wiązanie leków z
białkami jest też zmniejszone ze względu na obniżone
poziomy albumin i obecność albuminy płodowej, która
hamuje wiązanie leków [12, 13, 16]. Taka sytuacja prowadzi do utrzymywania się zwiększonych poziomów
leków niezwiązanych z białkami i wystąpienia objawów intoksykacji [12, 13, 15–18]. Metabolizm leków
u noworodków jest wyraźnie wolniejszy z powodu
zmniejszonej aktywności enzymów mikrosomalnych.
Normalizacja tego procesu jest zauważalna ok. 12 miesiąca życia [16, 17]. U noworodków procesy eliminacji
nerkowej są także zmienione na skutek niedojrzałości
czynnościowej nerek. Wartości Cl leków są znacznie
podwyższone [17]. U niemowląt przy poprawiającej
się perystaltyce, zwiększaniu się powierzchni wchłaniania i aktywności enzymów, proces wchłaniania leków poprawia się [16]. Wiązanie się leków z białkami
jest nadal obniżone [12, 13, 15]. Metabolizm wątrobowy z powodu małej jeszcze aktywności enzymatycznej
jest wolny. Stąd też wartości t½ są podwyższone [12,
13]. Dojrzewanie metabolizmu wątrobowego u dzieci polega na spowalnianiu tego procesu aż do okresu
dojrzewania [16]. Wydalanie nerkowe również ulega
11
Barbara Steinborn
zmianom, ale wartości Cl nadal są znacznie wyższe
niż w populacji dzieci starszych i dorosłych [18–21].
Wszystkie te odmienności powodują przede wszystkim
potrzebę częstszego podawania LPP, ale też w ten sposób zmniejsza się ryzyko występowania wahań dobowych stężeń leków w surowicy krwi i ewentualnych
objawów niepożądanych [17]. Stężenia FBM, VGB,
TGB, TPM i ZNS, a także konwencjonalnych LPP są
niższe w tej grupie wiekowej pacjentów [22].
Potrzeba znajomości stężeń
terapeutycznych – konieczność
monitorowania leczenia
Dla konwencjonalnych LPP (fenytoina – PHT, prymidon – PRM, fenobarbital – PB, karbamazepina –
CBZ, etosuksymid – ETH, kwas walproinowy – VPA),
wprowadzonych do leczenia do końca lat osiemdziesiątych ubiegłego stulecia, udowodniono występowanie
zależności pomiędzy stężeniem w surowicy a efektem
klinicznym, a także występowaniem objawów toksycznych. Zależność ta stała się podstawą do wprowadzenia tzw. terapii monitorowanej padaczki. Taki sposób prowadzenia leczenia tego zespołu chorobowego
pozwala na wykorzystanie znajomości stężeń LPP w
płynach biologicznych (surowica, ślina, łzy) i zapewnia przez: to możliwość zmiany sposobu dawkowania
LPP, unikanie objawów niepożądanych, zależnych od
poziomów tzw. potencjalnie toksycznych, prowadzenie leczenia w systemie politerapii, podawanie LPP
w różnych przedziałach wiekowych i współistnienie
innych zespołów chorobowych [23-29]. Wartość tej
metody postępowania w leczeniu padaczki była wielokrotnie dyskutowana na przestrzeni ostatnich 30-40
lat i została zaakceptowana przez Komisję LPP Międzynarodowej Ligi Przeciwpadaczkowej. Komisja ta
przedstawiła także wytyczne monitorowanego leczenia
padaczki [9]. Według Eadiego, monitorowane leczenie
padaczki pomaga w poznaniu błędów terapeutycznych
związanych z przedawkowaniem LPP lub częściej ze
stosowaniem dawek zbyt niskich lub podawaniem LPP
w odpowiednich dawkach, a mimo to braku kontroli napadów [30]. Ma to ułatwić decyzję o włączeniu
innych LPP. Pozwala także na identyfikację sytuacji, w których przedawkowanie LPP może zwiększyć
częstość napadów padaczkowych, jak również ocenić
niestosowanie się do zaleceń lekarskich oraz interakcje
farmakokinetyczne, które mogą ułatwić stosowanie odpowiedniej terapii.
Monitorowane leczenie padaczki jest uważane,
mimo upływu lat, za nowoczesny sposób postępowania [6, 9] przy założeniu, że prawidłowo zaplanowany
jest sposób pobierania krwi do badania i odpowiednio
interpretowany wynik pomiaru [6, 31]. Niewątpliwie
12
istotnym wskazaniem, poza oceną niepowodzeń w leczeniu i tzw. potencjalnej toksyczności (wynikającej
m.in. z wolnego metabolizmu i interakcji lekowych),
jest ustalenie poziomu wyjściowego i zmiana leczenia
LPP [6]. Terapia monitorowana padaczki została uznana w leczeniu padaczki także i w Polsce. Zespół ekspertów Komisji Polskiego Towarzystwa Epileptologii
wymienia monitorowanie stężeń LPP w standardach
postępowania w leczeniu padaczki i podaje wskazania
do tego rodzaju badań [32].
Konwencjonalne LPP spełniają warunki do prowadzenia monitorowanego leczenia ze względu na tzw.
wąski indeks terapeutyczny, dużą rozpiętość dawek terapeutycznych, co może być związane ze zróżnicowaniem indywidualnej farmakokinetyki, występowanie
efektu terapeutycznego po pewnym czasie od wprowadzenia leczenia oraz trudności w rozpoznaniu objawów
toksycznych tylko za pomocą oceny klinicznej [7, 33,
34].
Koncepcja monitorowanego leczenia nowymi LPP
nie została we wszystkich przypadkach jednoznacznie
określona. Może to być związane z prowadzeniem leczenia tymi lekami głównie w formie terapii dodanej
i trudnościami w jednoznacznym określeniu tzw. stężeń optymalnych. Nowe LPP charakteryzują się przede
wszystkim tzw. liniową kinetyką, co ułatwia przewidywanie efektów w zależności od zmian dawki z wyjątkiem GBP, gdzie obserwowano spadek absorpcji przy
wzroście dawki [35]. Poza tym nowe LPP praktycznie
nie indukują enzymów wątrobowych i rzadko wchodzą w interakcje lekowe [36, 37]. Po początkowym zachwycie i podkreślaniu braku wskazań do pomiarów
stężeń tych leków w płynach biologicznych, obecnie
dyskutuje się potrzebę prowadzenia badań farmakokinetycznych w różnych zespołach chorobowych i specjalnych populacjach pacjentów [34]. Dawkowanie
nowych LPP ustalono empirycznie, pomiary stężeń
leków przy tych dawkach stanowią użyteczną informację, która może być wykorzystana w leczeniu. Poza
tym pomiar w późniejszym okresie, np. przy wystąpieniu objawów niepożądanych, napadów padaczkowych
czy interakcji z innymi lekami, a także podejrzeniu o
niestosowanie się do zaleceń lekarskich, pozwala na
lepszą i pełniejsza ocenę stosowanych dawek. Znajomość stężeń leków ułatwia, co ważniejsze, ustalenie
dalszego dawkowania. Ta strategia sprawdziła się dla
leków konwencjonalnych i nie ma powodu, według Perucci, aby nie postępować tak również przy stosowaniu
nowych LPP [34].
Wśród własności farmakologicznych nowych LPP,
które umożliwiają prowadzenie monitorowanego leczenia, Tomson i Johannensen wymieniają następujące
cechy: wąski indeks terapeutyczny, występowanie zależności stężenie/efekt kliniczny lub objawy toksyczNeurologia Dziecięca
Znaczenie badań farmakokinetycznych leków przeciwpadaczkowych w leczeniu padaczki u dzieci i młodzieży
ne, indywidualne różnice w farmakokinetyce oraz jej
zmienność w zależności od wieku, interakcji lekowych
czy współistnienia dodatkowych chorób [38].
Znaczenie pomiarów poziomów
leków niezwiązanych
z białkami surowicy krwi
w leczeniu padaczki
Pomiary stężeń leków niezwiązanych z białkami,
które są aktywną frakcją, odpowiedzialną za efekt farmakologiczny, tzn. kliniczny i ewentualne pojawienie
się objawów niepożądanych, mają swoją ugruntowaną
pozycję w terapii monitorowanej [39]. Monitorowanie stężeń leków niezwiązanych z białkami były także
przedmiotem własnych, wcześniej publikowanych obserwacji [40, 41].
Przydatność kliniczna oznaczeń stężeń leków niezwiązanych z białkami została potwierdzona i jest
wykorzystywana w praktyce klinicznej [42], zwłaszcza gdy zmiany tych stężeń mogą mieć konsekwencje
terapeutyczne i toksyczne [18]. Zasada ta odnosi się
szczególnie do leków wykazujących wysoki odsetek
wiązania się z białkami surowicy krwi.
Wśród konwencjonalnych LPP są to PHT, VPA i
CBZ. Dla PHT wiązanie się z białkami surowicy krwi
wynosi 75–95%, dla CBZ 75–80% a dla VPA 90% [3,
43]. Jeśli dodatkowo zaistnieją okoliczności prowadzące do zmian ich wiązania się z białkami, to wskazania
do ich pomiarów są tym bardziej uzasadnione [42].
Najczęściej wymienianymi wskazaniami do takich
badań są stany prowadzące do zmniejszenia się poziomów białek, tzn. hipoalbuminemia zarówno w fizjologii (ciąża, wiek podeszły), jak i w niewydolności nerek,
przy utracie krwi i oparzeniach [9].
Szczególnie często podkreśla się potrzebę oznaczania stężeń leków niezwiązanych w przypadkach niewydolności nerek i chorób wątroby [9, 44]. Wzrosty
stężeń wolnego VPA z jednoczesnymi objawami niepożądanymi odnotowano także po transplantacji serca i
operacjach neurochirurgicznych, co może być związane ze spadkami stężenia albumin i utratą krwi. Zmiany
stężeń niezwiązanych leków występują przy kojarzeniu leków ze sobą, zwłaszcza takich, które wykazują
zdolność wypierania innych z połączeń białkowych.
[44-46]
Zmiany wiązania się z białkami surowicy krwi
mogą więc wywoływać wystąpienie objawów niepożądanych, tj. toksycznych, a także być przyczyną zmian
efektów tłumienia napadów padaczkowych [47]. Zmiana wiązań z białkami może nie dawać w efekcie obniżenia poziomu całkowitego leku, stąd pomiar tylko
całkowitej ilości leku może nie być obrazem zachodzącej interakcji. Jeśli jeden lek wypiera z połączeń
Vol. 15/2006, Nr 29
z białkami drugi, to dochodzi do znacznego wzrostu
poziomu leku niezwiązanego [48]. Przykładem takiej
interakcji może być połączenie niesteroidowych leków
przeciwzapalnych, w tym aspiryny, z VPA. Podczas
jednoczesnego podawania tych preparatów opisywano
szereg objawów niepożądanych, związanych ze zwiększonym poziomem VPA [49, 50].
Kolejnym przykładem takiej interakcji jest biterapia
PHT z VPA. VPA wypiera PHT z jej połączeń z białkami, a jednocześnie hamuje jej metabolizm, obniżając
całkowity poziom w surowicy krwi, a poziom niezwiązanej PHT może nie zmienić się [51]. Dla prawidłowego zaplanowania dawkowania PHT w interakcji z VPA
konieczna jest więc ocena obu stężeń PHT w surowicy
krwi.
Stosując preparaty VPA w różnych grupach wiekowych i oceniając wzajemne proporcje stężenia jego niezwiązanej frakcji z całkowitym stężeniem, zauważono,
że stężenie wolne rośnie nieliniowo wraz ze wzrostem
całkowitego stężenia VPA, jako efekt wysycania się
wiązań z białkami [1, 52]. Obserwuje się jednak znaczne różnice międzyosobnicze [12].
Wiązanie CBZ z białkami według Battino i wsp.
zależy od wielu czynników: interakcji zachodzących
na poziomie wiązania się z białkami, wieku, czasu pobierania próbek oraz dodatkowego leczenia [13, 53]. Z
białkami surowicy krwi wiąże się także aktywny metabolit CBZ-E, ale jego powinowactwo do białek jest
ok. 4 razy słabsze niż leku głównego. To właśnie zmiany wiązania się tego metabolitu po włączeniu innych
leków są opisywane częściej niż leku głównego [13,
54].
Pomiary stężeń leków niezwiązanych z białkami
przeprowadza się metodami takimi, jak i leków całkowitych przy wcześniejszym wykorzystaniu zestawów do ultrafiltracji, zapewniających odseparowanie
części leku niezwiązanej z białkami. Alternatywą dla
oznaczeń stężeń leków niezwiązanych z białkami są
ich badania w ślinie, co prowadzono także w ramach
wcześniejszych badań własnych [26]. Nie wszystkie
jednak leki mogą być w ten sposób monitorowane.
Szczególnie mało przydatne jest monitowanie VPA w
ślinie [26].
Podsumowanie
Ze względu na odmienności farmakokinetyki LPP u
dzieci przydatna w leczeniu pacjentów tej grupy wiekowej wydaje się przede wszystkim znajomość okresu
biologicznego półtrwania i zakresu terapeutycznego
stężeń oraz dróg eliminacji. Dla oceny skuteczności
i możliwych objawów niepożądanych ważna jest też
znajomość potencjalnych interakcji LPP.
13
Barbara Steinborn
Piśmiennictwo
[1] Cloyd J. C., Remmel R. R.: Antiepileptic drug pharmacokinetics and interactions: imapact on treatment of epilepsy.
Pharmacotherapy, 2000: 20, 8, 139.
[2] Riva R. et al.: Pharmacokinetic interaction between antiepileptic drugs.Clinical consideration. Clin. Pharmacokinet.,
1996: 31, 6, 470.
[3] Faught E.: Pharmacokinetic consideration in prescribing antiepileptic drugs. Epilepsia, 2001: 42, 4, 19.
[4] Spear B. B.: Pharmacogenetics and antiepileptic drugs. Epilepsia, 2001: 42, 5, 31.
[5] Browne T. R.: Pharmacokinetics of antiepileptic drugs. Neurology, 1998: 51, 4, 2.
[6] Glauser T., Pippenger C. E.: Controversies in blood-level monitoring: Reexamining its role in treatment of epilepsy.
Epilepsia, 2000: 41, 8, 6.
[7] Bourgeois B. F.: Pharmacokinetic properties of current antiepileptic drugs. What improvements are needed? Neurology,
2000: 55, 3, 11.
[8] Patsalos P. N.: Pharmacokinetic profile of levetiracetam: toward ideal characteristics. Pharmacol. Ther., 2000: 85, 2,
77.
[9] Commission on Antiepileptic Drugs, ILAE: Guidelines for therapeutic monitoring of antiepileptic drugs. Epilepsia,
1993:34,585.
[10] Rowan A. J. et al.: The delayed effect of sodium valproate on the photoconvulsive response in man. Epilepsia, 1979:
20, 61.
[11] Ferrendelli J. A.: Concerns with antiepileptic drug initiation: safety, tolerability, and efficacy. Epilepsia, 2001: 42, 4,
28.
[12] Battino D., Estienne M., Avanzini G.: Clinical pharmacokinetics of antiepileptic drugs in paediatric patients. Part I: phenobarbital, primidone, valproic acid, ethosuximide and methosuximide. Clin. Pharmacokinet., 1995: 29, 4, 257.
[13] Battino D., Estienne M., Avanzini G.: Clinical pharmacokinetics of antiepileptic drugs in paediatric patients. Part II:
phenytoin, carbamzepine, sulitame, lamotrigine, vigabatrin, oxcarbazepine and felbamate. Clin. Pharmacokinet., 1995:
29, 6, 341.
[14] Anderson G. et al.: Time course of lamotrigine de-induction: impact of step-wise withdrawal of carbamazepine or phenytoin. Epilepsy Res., 2002: 49, 3, 211.
[15] Anderson G. D.: Children versus adults: pharmacokinetic and adverse-effect diffrerences. Epilepsia, 2002: 43, 3, 53.
[16] Sobaniec W., Kułak W., Sobaniec-Łotowska M.: Farmakokinetyka leków przeciwpadaczkowych u dzieci. Neur. Dziec.,
1993: 2, 3, 77.
[17] Sobaniec W., Sobaniec H., Sobaniec-Łotowska M.: Monitorowane leczenie padaczki u dzieci [w]: Postępy w diagnostyce i leczeniu chorób układu nerwowego u dzieci. Tom 2. Red. Jóźwiak S., Bifoilum, Lublin, 2000.
[18] Herngren L., Lundberg B., Nergardh A.: Pharmacokinetics of total and free valproic acid during monotherapy in infants.
J. Neurol., 1991: 238, 315.
[19] Herngren L., Nergardh A.: Pharmacokinetics of free and total sodium valproate in adolescents and young adults during
maintenance therapy. J. Neurol., 1988: 235, 491.
[20] Cloyd J. C. et al.: Valproic acid pharmacokinetics in children. IV. Effects of age and antiepileptic drugs on protein binding and intristic clearence. Clin. Pharmacol. Ther., 1993: 53, 1, 22.
[21] Sundquist A., Tomson T., Lundkvist B.: Pharmacokinetics of valproic acid in patients with juvenile myoclonic epilepsy
on monotherapy. Ther. Drug. Monit., 1997: 19, 153.
[22] Perucca E.: The clinical pharmacokinetics of the new antiepileptic drug. Epilepsia, 1999: 40, 9, 7.
[23] Jannuzzi G., Cian P., Fattore C.: A multicenter randomized controlled trial on the clinical impact of therapeutic drug
monitoring in patients with newly diagnosed epilepsy. The Italian TDM study group in epilepsy. Epilepsia, 2000: 41,
2, 222.
[24] Mattson R. H.: Antiepileptic drug monitoring. A reappraisal. Epilepsia, 1995: 36, 5, 22.
[25] Snodgrass S. R., Parks B. R.: Anticonvulsant blood levels: historical review with a pediatric focus. J. Child. Neurol.,
2000: 15, 734.
[26] Steinborn B.: Korelacja między stężeniami leków przeciwpadaczkowych w ślinie i surowicy krwi u dzieci. Probl. Ter.
Monit., 1995: 6, 2, 60.
[27] Steinborn B., Galas-Zgorzalewicz B.: Monitorowanie stężeń leków przeciwpadaczkowych – trzynaście lat doświadczeń
w Pracowni Neurofarmakologii Klinicznej Katedry i Kliniki Neurologii Wieku Rozwojowego. Neur. Neurochir. Pol.,
2000: 1, 49.
[28] Steinborn B.: Monitorowane leczenie padaczki przy stosowaniu nowych leków przeciwpadaczkowych. Probl. Ter.
Monit., 2003: 14, 1, 19.
[29] Yukawa E.: Optimisation of antiepileptic drug therapy. The importance of serum drug concentration monitoring. Clin.
Pharmacokinet., 1996: 31, 2, 120.
[30] Eadie M. J.: The role of therapeutic drug monitoring in improving the cost effectiveness of anticonvulsant therapy. Clin.
Pharmacokinet., 1995: 29, 1, 29.
14
Neurologia Dziecięca
Znaczenie badań farmakokinetycznych leków przeciwpadaczkowych w leczeniu padaczki u dzieci i młodzieży
[31] Majkowski J.: Terapeutyczne monitorowanie leków przeciwpadaczkowych w surowicy: ograniczenia i zalety. Epileptologia, 1999: 7, 343.
[32] Majkowski J.: Standardy diagnostyki i leczenia chorych z padaczką w Polsce. Epileptologia, 2002: 10, 2, 109.
[33] Steinhoff B. J. et al.: The ideal characteristics of antiepileptic therapy: an overview of old and new AEDs. Acta Neurol.
Scand., 2003: 107, 87.
[34] Perucca E.: Is there a role for therapetic drug monitoring of new anticonvulsants? Clin. Pharmacokinet., 2000: 38, 3,
191.
[35] Perucca E.: The clinical pharmacokinetics of the new antiepileptic drugs. Epilepsia, 1999: 40, 9, 7.
[36] Bialer M. et al.: Progress report on new antiepileptic drugs: a summary of the Sixth Eilat Conference (EILAT VI). Epilepsy Res., 2002: 51, 1-2, 31.
[37] Perucca E.: Marketed new antiepileptic drugs. Are they better than old-generation agents? Ther. Drug Monit., 2002: 24,
74.
[38] Tomson T., Johannesen S. I.: Therapeutic monitoring of the new antiepileptic drugs. Eur. J. Clin. Pharmacol., 2000: 55,
10, 697.
[39] Lenn N. J., Robertson M.: Clinical utility of unbound antiepileptic drug blood levels in the management of epilepsy.
Neurology, 1992: 42, 988.
[40] Steinborn B.: Przydatność kliniczna oznaczeń stężenia kwasu walproinowego i jego wolnej frakcji w surowicy podczas
leczenia padaczki u dzieci. Probl. Ter. Monit., 1991: 2, 15.
[41] Steinborn B., Galas-Zgorzalewicz B.: Clinical and pharmacokinetic obsrevations on valproate: an 3 year follow-up study
in 100 children with generalised epilepsy. Funct. Neurol., 1993: 8, 6, 415.
[42] Soldin S. J.: Free drug measurements. Arch. Pathol. Lab. Med., 1999: 123, 822.
[43] Guberman A., Bruni J.: Essential of clinical epilepsy. Butterworth Heinemann, Boston 1999.
[44] Dasgupta A., Jacques M.: Reduced in vitro displacement of valproic acid from protein binding by salicylate in uremic
sera compared with normal sera. Role of uremic compounds. Am. J. Clin. Pathol., 1994: 101, 349.
[45] Haroldson J. A. et al.: Elevated free of valproic acid in a heart transplant patient with hypoalbuminemia. Ann. Pharmacother., 2000: 34, 183.
[46] Leiri L. et al.: Pharmacokinetic study of valproic acid sustained – release preparation in patients undergoing brain surgery. Ther. Drug Monit., 1995: 17, 6.
[47] Kodama Y. et al.: Comparison of two binding equations for prediction of the concentration of unbound valproic acid in
the serum of adult epileptic polytherapy patients. J. Pharm. Pharmacol., 1996: 48, 1068.
[48] French J. A., Gidal E. B.: Antiepileptic drug interaction. Epilepsia, 2000: 41, 8, 30.
[49] Dasgupta A., Emerson L.: Interaction of valproic acid with nonsteroidal antiinflammatory drugs mefenamic acid and
fenoprofen in normal and uremic sera: lack of interaction in uremic sera due to the presence of ednogenus factors. Ther.
Drug Monit., 1996: 18, 6, 654.
[50] Dasgupta A., Volk A.: Displacemnet of valproic acid and carbamazepine from protein binding in normal nad uremic
sera by tolmentin, ibuprofen and naproxen; presence of inhibitor in uremic serum that blocks valproic acid-naproxen
inetraction. Ther. Drug Monit., 1996: 18, 284.
[51] Patsalos P. N.: The importance of drug interactios in epilepsy therapy. Epilepsia, 2002: 43, 4, 365.
[52] Cloyd J. C. et al.: Valproate unbound fraction distribution volume following rapid infusion in patients with epilepsy.
Epilepsy Res., 2003: 53, 19.
[53] Kodama H. et al.: Age-related alteration of carbamazepine-serum protein binding in man. J. Pharm. Pharmacol., 1999:
51, 9, 1009.
[54] Kodama H., Tsutsumi K., Kuranari M.: In vivo binding characteristics of carbamazepine and carbamazepine 10, 11-epoxide to serum proteins in paediatric patients in epilepsy. J. Clin. Pharmacol., 1993: 44, 3, 291.
Adres autora:
Katedra i Klinika Neurologii Wieku Rozwojowego
Akademia Medyczna im. Karola Marcinkowskiego
60-355 Poznań
ul. Przybyszewskiego 49
Vol. 15/2006, Nr 29
15