Genetyka nowotworów
czynniki środowiskowe i genetyczne
choroba genetyczna dotycząca komórek
somatycznych
1/3 osób w populacji
nowotwory dziedziczne 5 % częstych
nowotworów (rak sutka, rak jelita grubego)
predyspozycja genetyczna dla jednego
rodzaju nowotworu
rodzinny zespół predyspozycji do rozwoju
różnych nowotworów
proces wieloetapowy - kumulacja mutacji wielu
genów komórkowych - klonalna selekcja komórek
charakteryzujących się agresywnym wzrostem
mutacje trzech klas genów: onkogenów, genów
supresorowych i genów naprawczych
nowotwór jest następstwem sumowania się skutków
mutacji.
pierwsza mutacja może być odziedziczona
(nowotwory rodzinne), zwykle powstaje w komórkach
somatycznych wraz z upływem czasu
czas niezbędny do skumulowania skutków mutacji
45-letnia kobieta diagnozowana z powodu
powiększonej śledziony, L:31 tys, płytki:650 tys, szpik
bogatokomórkowy
w badaniu cytogenetycznym w wielu komórkach
szpiku obecny tzw. chromosom Philadelphia (95%
osób)
der(22)t(9;22)(q34;q11.2)
rozpoznanie: przewlekła białaczka szpikowa
klonalny rozrost komórek układu hematopoetycznego
szpiku
nadmierna ekspresja onkogenu BCR-ABL
ABL (protoonkogen Abelson) kodujący kinazę
tyrozynową (9q34-udział w cyklu komórkowym i
przekazywaniu sygnałów)
BCR (gen regionu breakpoint cluster) koduje
fosfoproteinę (22q11)
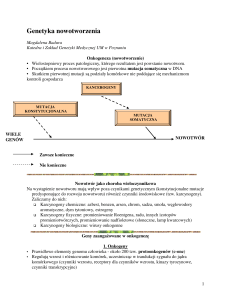
Protoonkogeny
RET
kinaza tyrozynowa
mutacje
punktowe
MYC
cz. transkrypcyjny
translokacje
reguluje cykl i
amplifikacje
aktywność telomerazy mutacje
cz.transkrypcyjny
mutacje
katenina
BCL2 hamuje apoptozę
translokacje
ABL
reguluje cykl
translokacje
MDM2 inaktywacja p53
amplifikacja
K-RAS przekazywanie
N-RAS sygnału
H-RAS
mutacje
MEN2a i b
rak brodawczakowaty
tarczycy
chłoniak
Burkitta
polipowatość jelita
grubego
chłoniaki
CML
mięsaki kości,
tk.miękkich
60-80% raków trzustki
25-30% różnych guzów
2-letni chłopiec: zez zbieżny prawego oka,
leukokoria (biały refleks źrenicy), w badaniu
okulistycznym pojedynczy guz w obrębie
siatkówki.
Retinoblastoma guz embionalny 1/18-30 tys.
gen RB1 gen supresorowy guzów
białko Rb -reguluje procesy proliferacji komórek
w czasie cyklu komórkowego
Teoria dwóch uderzeń („two hits”)
40% mutacja germinalna (ale tylko 10% ma
wywiad)
nosiciele ryzyko innych nowotworów: kostniakomięsaki, mięsaki tkanek miękkich, czerniaki
postać obustronna, wieloogniskowa (30%)
objawy w 1 r.ż.
Postać jednostronna (70%) objawy 24-30 m.ż.
30% ma mutację germinalną
pacjenci z postacią wrodzona wymagają opieki do
7 r.ż
badania molekularne: ocena mutacji w tkance
guza, poszukiwanie tych mutacji we krwi pacjenta
osoba z postacią obustronną 50% szans na
przekazanie genu potomstwu, rozwoju choroby
45% (bo bardzo duże ryzyko mutacji
somatycznej)
osoba z postacią jednostronną ryzyko 7-15%
(może mieć mutację)
90% dzieci z retinoblastoma to pierwsze osoby w
rodzinie
!! Badać rodziców okulistycznie.
27-letnia kobieta zgłasza się do poradni
genetycznej, rozpoznano u niej raka sutka,
zarówno jej matka jak i dwie ciotki ze strony matki
i dziadek ze strony matki mieli raka sutka.
Wykonano badania w kierunku mutacji BRCA1 i
BRCA2. Jest nosicielka mutacji BRCA2.
Po wykonaniu badań u członków rodziny, jedna
kobieta zdecydowała się na profilaktyczna
mastektomię jedna na usuniecie jajników.
Mutacje tych dwóch genów w 70-80% rodzinnych
raków sutka
BRCA1 i BRCA2 kodują białka jądrowe
utrzymujące integralność genomu poprzez
regulację naprawy DNA i cyklu komórkowego.
Geny supresorowe, tworzenie guza według teorii
„dwóch uderzeń”, somatyczna utrata drugiego
allelu.
BRCA1 i BRCA2 predysponują do raka sutka i
jajnika
BRCA1 - rak prostaty, jelita grubego
BRCA2 - rak prostaty, trzustki, dróg żółciowych,
pęcherzyka żółciowego i raka sutka u mężczyzn
ryzyko odziedziczenia 50% ale niekompletna
penetracja i różna ekspresja nie można
przewidzieć wieku wystąpienia
brak odziedziczonej mutacji nie przesądza o
mniejszej predyspozycji, krewni I stopnia mogą
mieć wspólne geny o mniejszej penetracji
Kumulacyjne ryzyko w wieku 70 lat (%)
BRCA1
BRCA2
populacja
rak sutka
jajnika
prostaty
40-87
28-84
8-10
16-63
27
1.5
25
20
10
Empiryczne ryzyko rozwoju raka sutka
populacyjne
1/10-12
8%
krewna I >55r.ż.
1/8
12%
krewna I <55 r.ż.
1/6
18%
krewna I <45 r.ż.
1/3
30%
krewna I z obustr.
1/2
50%
Rak sutka profilaktyka
Gdy ryzyko 2x populacyjne (1/6):
co roku badanie przedmiotowe
mammografia od 35 r.ż. co roku do 50 r.ż
potem co 18 m-cy
biopsja cienkoigłowa
badani nad tamoxifenem (antyestrogen)
usunięcie jajników u nosicieli BRCA1
zabezpiecza przede rakiem jajnika i zmniejsza
ryzyko raka sutka o 50% mastektomia (ryzyko
1/4) ??
50% mieszkańców krajów zachodnich do 70r.ż. będzie mieć
guza jelita grubego z tego 10% rozwinie się rak.
35-letni pacjent po całkowitej kolektomii skierowany do
poradni genetycznej ( w badaniu mnogie polipy, hist-pat
polipowatość jelita grubego), w badaniu dna oka ma
wrodzony przerost nabłonka barwnikowego siatkówki.
Wielu krewnych zmarło z powodu raka jelita grubego.
Wykryto mutacje genu APC.
FAP (rodzinna polipowatość jelita grubego, APC)
2-3/10 tys stanowi 1% raków jelita grubego
15% raków jelita grubego ma charakter rodzinny
mutacje APC stwierdza się w 80% guzów sporadycznych
Apc reguluje transkrypcję, adhezje komórkową, migracje
komórek, apoptozę i proliferację.
Proces rozwoju raka jelita grubego proces
wieloetapowy.
Poza mutacja genu APC dodatkowo aktywacja
onkogenów K-ras, N-ras, inaktywacja supresora
na 18q, inaktywacja p53 itd.
W trakcie gromadzenia mutacji -komórki
nabywają cech inwazyjnych
FAP - -setki polipów w jelicie grubym pomiędzy 740 r.ż. penetracja 7% do 21r.ż., 87% do 45 r.ż., 93%
do 50 r.ż.
CHRPE (congenital hypertrophy of retinal
pigment epithelium)
niepełna choć prawie całkowita penetracja
20-30% pacjentów to nowe mutacje, różna
ekspresja u członków rodzin, nie można
przewidzieć nasilenia zmian czy wystąpienia
choroby
Profilaktyka
sigmoidoskopia co 1-2 lata od 10-12 r.ż.
Zespoły alleliczne:
zespół Gardnera: kostniaki żuchwy, torbiele
naskókowe
zespół Turcota: pierwotne guzy mózgu
Geny supresorowe
p53
cz.transkrypcyjny zespół Li-Fraumeni, większość
cykl komórkowy
guzów sporadycznych
apoptozę
Rb
kontrola przejścia
do fazy S
retinoblastoma, guzy
sporadyczne
VHL
hamuje czynnik wzrostu
środbłonka naczyń
zespól von Hippel-Lindau
(liczne hemangioma)
WT1
czynnik transkrypcyjny
wpływ na ekspresję p53
dziedziczny nefroblastoma
guz Wilmsa
NF1
inaktywuje aktywną
postać onkogenu RAS
neurofibromatoza typ1
czynnik transkrypcyjny
guzy sporadyczne, młodzieńcza
polipowatość jelita
BRCA1
SMAD4
38 -letnia kobieta skierowana do poradni genetycznej z
powodu wywiadu rodzinnego: ojciec, brat, wielu krewnych
II miało raka jelita grubego a siostra i ciotka dodatkowo
raka endometrium.
Wywiad sugeruje HNPCC (hereditary nonpolyposis colon
cancer) niepolipowaty rak jelita grubego
w badaniu guza u osoby chorej poszukuje się zjawiska
niestabilności powtórzeń mikrosatelitarnych potem mutacji
w genach naprawczych
HNPCC mutacje w genach naprawczych 2-5/1000
3-8% wszystkich raków jelita grubego
geny naprawcze: MSH2,MSH6, MLH1, MLH3, PMS1,PMS2
(homologi genów mutatorowych u E. Coli) geny naprawcze
błędnie sparowanych zasad
Błędy w syntezie prowadzą do deformacji podwójnej helisy
DNA co przyciąga białka kodowane przez geny mutatorowe
a takie kompleksy przyciągają inne enzymy służące do
naprawy
HNPCC 80% penetracji, średni wiek <50 r.ż.
brak polipów, podejrzenie tylko na podstawie wywiadu
inne nowotwory w rodzinach: rak żołądka, jelita cienkiego,
trzustki, nerki, endometrium, jajnika
nie ma ryzyka raka sutka czy płuc
profilaktyka kolonoskopia od 25 r.ż., kolektomia
ryzyko przekazania genu 50%, rozwoju choroby 45%
niekompletna penetracja, zróżnicowana ekspresja
nie można przewidzieć ani wieku wystąpienia ani nasilenia
objawów
Wywiad:
wykluczenie FAP
3 członków rodziny z taka samą diagnozą
jeden jest krewnym I dwóch pozostałych
choroba w co najmniej dwóch pokoleniach
przynajmniej u jednej osoby diagnoza <50 r.ż.
Rozróżnia się
Lynch 1- nowotwory jelita grubego
Lynch 2- dodatkowo inne nowotwory
Inne defekty naprawy DNA
Ataksja teleangiektazja:
defekt zahamowania komórki po uszkodzeniu
DNA: uszkodzenie móżdżku, upośledzenie
odporności, wrażliwość na promieniowanie,
niestabilność chromosomów, predyspozycja do
białaczek i chłoniaków
Zespól Blooma:
niedobór helikazy uczestniczącej w replikacji
DNA: niski wzrost, wrażliwość na światło,
upośledzenie odporności, niestabilność
chromosomów, predyspozycja do nowotworów
Xeroderma pigmentosum:
upośledzona naprawa DNA: nadwrażliwośc na
światło słoneczne, plamy piegowate na skórze,
czerniaki
Cechy sugerujące dziedziczną
predyspozycję do nowotworów
Wielu krewnych I lub II stopnia z
powszechnymi nowotworami
wielu krewnych z tzw. pokrewnymi
nowotworami: sutka i jajnika, jelita grubego i
endometrium
dwóch członków z takim samym rzadkim
nowotworem
nietypowy bardzo wczesny wiek wystąpienia
obustronny nowotwór
nawrót nowotworu
nowotwór dotyczący dwóch różnych narządów
u tej samej osoby