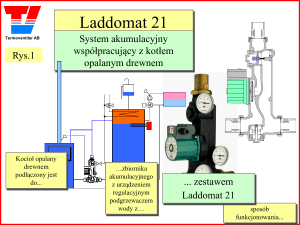
Analiza czynników głównych i inne metody eksploracji danych
M. Daszykowski i B. Walczak
Zakład Chemometrii, Instytut Chemii, Uniwersytet Śląski, ul. Szkolna 9, 40-006 Katowice
http://www.chemometria.us.edu.pl
1. WPROWADZENIE
Proces badawczy jest zwykle procesem wieloetapowym. Składa się na niego
planowanie eksperymentu, pobranie próbek, analiza chemiczna, kontrola jakości
uzyskanych danych, ich chemometryczna analiza i interpretacja [1]. W niniejszym
rozdziale, skupimy się jedynie na analizie danych, omawiając, w sposób ogólny i
możliwie przystępny, niektóre techniki chemometryczne stosowane do eksploracji
wielowymiarowych danych chemicznych.
Obecnie, w wielu problemach analitycznych dane uzyskuje się jako rezultat analiz
szeregu próbek. Wyniki analiz można zorganizować w macierz danych, X, gdzie m
wierszy macierzy odpowiada m mierzonym próbkom, a n kolumn odpowiada n
mierzonym parametrom. Schematycznie, macierz danych przedstawiono na Rys. 1a.
W zależności od stosowanej techniki lub technik analitycznych do opisu badanej
próbki lub układu fizyko-chemicznego, wiersze macierzy danych mogą tworzyć
sygnały instrumentalne (np. widma UV-VIS zmierzone w określonym zakresie
spektralnym, chromatogramy, widma masowe, etc.) lub wektory, o elementach
reprezentujących wyniki n analiz (np. stężenia elementów śladowych w próbce,
stężenia wybranych kwasów tłuszczowych, etc.).
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
1
n
parametry
n
próbki
a)
m
macierz danych X[m,n]
n
b)
X
=
X*
+
X~
m
dane analityczne
prawdziwy sygnał analityczny
błąd pomiarowy
Rys. 1 a) Graficzne przedstawienie macierzy danych o m wierszach (nazywanych obiektami lub
próbkami) i n kolumnach (nazywanych zmiennymi lub parametrami), b) poszczególne składowe
macierzy danych X: prawdziwy sygnał analityczny i błąd pomiarowy.
Każde dane analityczne obarczone są błędem pomiarowym, dlatego macierz danych
możemy przedstawić jako sumę dwóch komponentów, co pokazano na Rys. 1b.
Często zamiast słowa próbki używa się terminu obiekty, gdyż kolejne wiersze
macierzy mogą zawierać pomiary dla tej samej próbki w różnych odstępach czasu,
aby zaobserwować zachodzące w niej zmiany. Natomiast kolumny macierzy danych
nazywa się zmiennymi lub parametrami.
Rozważmy zbiór danych, który uzyskano oznaczając w 10 próbkach stężenia jonów
cynku i wapnia (zob. Rys. 2a). Macierz danych, X, ma wymiary 10×2.
Każda próbka, opisana n parametrami, to punkt w n wymiarowej przestrzeni
parametrów, a każdy parametr, to punkt w m wymiarowej przestrzeni próbek.
Podobieństwa pomiędzy poszczególnymi próbkami można analizować w przestrzeni
parametrów, a pomiędzy parametrami w przestrzeni próbek. Ponieważ, w przypadku
omawianych danych, każdą próbkę opisują jedynie dwa parametry, dlatego dane
możemy zwizualizować. Jednym ze sposobów jest przedstawienie próbek w
dwuwymiarowym układzie współrzędnych, którego osie tworzą dwa parametry, tj.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
2
stężenie jonów cynku i wapnia, co pokazano na Rys. 2b. Dwie próbki są do siebie
podobne, jeśli na projekcji znajdują się blisko siebie. Innymi słowy oznacza to, iż
różnice pomiędzy odpowiednimi wartościami parametrów są małe.
W przypadku parametrów, które opisane są wynikami pomiarów dla 10 próbek,
możliwa jest jedynie prezentacja projekcji parametrów, na płaszczyznę, zdefiniowaną
dwoma obiektami. Taką przykładową projekcję parametrów na płaszczyznę
zdefiniowaną przez próbki 1 i 2 przedstawiono na Rys. 2c.
próbki
1
2
1
4,7402 15,3603
2
3,8774 13,9617
3
4,3282 14,9011
4
4,1832 14,5799
5
4,6696 15,7262
6
4,5145 15,2296
7
4,1478 14,6528
8
3,6222 12,8837
9
4,5857 15,1654
10
4,1336 14,2203
b)
16
5
15.5
stężenie jonów wapnia
parametry
6
3
7
4
14.5
10
14
2
13.5
13
8
12.5
3.4
3.6
3.8
4
4.2
4.4
4.6
4.8
5
stężenie jonów cynku
c)
stężenie jonów wapnia
14
12
10
8
6
4
macierz danych X[m,n]
1
9
15
próbka 2
a)
stężenie jonów cynku
2
0
0
2
4
6
8
10
12
14
16
próbka 1
Rys. 2 a) Macierz danych, X, zawierająca 10 próbek i 2 parametry (odpowiednio stężenia jonów cynku
i wapnia), b) projekcja próbek na płaszczyznę zdefiniowaną przez parametry 1 i 2 oraz c) projekcja
parametrów na płaszczyznę zdefiniowaną przez próbki 1 i 2.
Zazwyczaj, jako miarę podobieństwa między dwiema próbkami używa się odległość
euklidesową [ 2 ]. Odległość euklidesowa między dwiema próbkami, p i q, w
przestrzeni n parametrów, określa następujący wzór:
d (p, q) =
n
∑(p
i =1
i
− qi )
2
(1)
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
3
Podobieństwa pomiędzy wszystkimi obiektami macierzy X można przedstawić za
pomocą macierzy odległości, D. Jest ona kwadratowa (o wymiarze m×m) i
symetryczna, ponieważ d(p,q) = d(q,p), a każdy jej element to odległość euklidesowa
obliczona pomiędzy i-tym, a j-tym obiektem macierzy X. Dla przedstawionych na
Rys. 2a danych, macierz odległości ma wymiary 10×10, a jej elementy przedstawiono
na Rys. 3.
0
1,6433
1,6433 0,6169 0,9587 0,3726 0,2608 0,9227 2,7172 0,2487 1,2913
0
1,0419 0,6896 1,9341 1,4189 0,7421 1,1077 1,3966 0,364
0,6169 1,0419
0
indeks obiektu
0,9587 0,6896 0,3524
0,3524 0,8929 0,3776 0,3069 2,1373 0,3689 0,708
0
0,3726 1,9341 0,8929 1,2452
1,2452 0,7292 0,081 1,7865 0,7105 0,363
0
0,2608 1,4189 0,3776 0,7292 0,5202
0,5202 1,1935 3,0293 0,567 1,5984
0
0,9227 0,7421 0,3069 0,081 1,1935 0,6834
0,6834 2,5098 0,0958 1,0787
0
2,7172 1,1077 2,1373 1,7865 3,0293 2,5098 1,8455
1,8455 0,6741 0,4327
0
0,2487 1,3966 0,3689 0,7105 0,567 0,0958 0,6741 2,4767
1,2913 0,364
0,708
2,4767 1,431
0
1,0476
0,363 1,5984 1,0787 0,4327 1,431 1,0476
0
indeks obiektu
Rys. 3 Macierz odległości euklidesowych, D, obliczonych dla wszystkich par
próbek macierzy danych X z Rys. 2a.
Z analizy macierzy odległości wynika, iż próbki 4 i 7 są najbardziej do siebie podobne,
a najbardziej różne, są próbki 5 i 8 (zob. Rys. 2b i 3).
W równaniu 1, kluczową rolę odgrywają wkłady różnic pomiędzy poszczególnymi
parametrami. Jeśli parametry są mierzone w różnych jednostkach i/lub mają różne
zakresy zmienności wówczas badanie podobieństw pomiędzy próbkami na podstawie
odległości euklidesowej nie prowadzi do poprawnych wniosków.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
4
Miarą zmienności parametru, mierzonego dla m badanych próbek, jest wariancja,
która również wyraża jego zawartość informacyjną:
∑ (x
m
var(x ) =
i =1
i
−x
)
2
(2)
m −1
Zmienne o wariancji bliskiej zeru nic nie wnoszą do opisu zróżnicowania danych i
dlatego takie zmienne można usunąć z danych. Wariancja jest wielkością addytywną.
Całkowitą wariancję danych wyrażamy jako sumę wariancji poszczególnych
zmiennych. Jednakże, wariancja nie jest wyrażona w tej samej jednostce, co dany
parametr, ze względu na obliczane kwadraty różnic pomiędzy elementami zmiennej, a
jej wartością średnią. Pierwiastek z wariancji, czyli odchylenie standardowe, ma tą
samą jednostkę co dana zmienna. W przypadku omawianych danych wariancje
parametrów wynoszą odpowiednio 0,1286 i 0,6786, a ich odchylenia standardowe
0,3586 i 0,8238.
W celu porównania dwóch parametrów, xk i xl, można użyć kowariancji, która
ilościowo określa ich liniową zależność [2]. Kowariancja dwóch parametrów
przyjmuje wartości z przedziału od -∞ do +∞. Dodatnie wartości kowariancji
świadczą o dodatniej ich zależności, a ujemne, o ujemnej:
∑ (x
m
cov(x k , x l ) =
i =1
ik
)(
− x k xil − x l
m −1
)
(3)
W przypadku parametrów macierzy danych X (zob. Rys. 2a) ich kowariancja jest
dodatnia i wynosi 0,2844. Wadą tej miary podobieństwa jest jej zależność od skali w
jakiej wyrażane są pomiary. Na przykład, podanie stężeń parametrów w ng⋅g-1
zamiast µg⋅g-1 zwiększa wartość obliczonej kowariancji o faktor 103. Z tego powodu,
w celu porównania dwóch parametrów zmierzonych w różnych jednostkach i/lub
różnej skali, stosuje się tzw. współczynnik korelacji Pearsona, gdyż pozwala on na
porównanie parametrów w różnych jednostkach i skalach. Usuniecie efektu różnych
jednostek i skal zmiennych uzyskuje się poprzez ich standardyzację. Operacja ta na
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
5
podzieleniu wszystkich elementów danej zmiennej przez jej odchylenie standardowe.
Po autoskalowaniu, odchylenie standardowe zmiennej i jej wariancja są jednostkowe.
r (x k , x l ) =
cov(x k , x l )
(4)
var(x k ) ⋅ var(x l )
Współczynniki korelacji przyjmują wartości pomiędzy -1, a 1. Duża wartość
współczynnika korelacji świadczy o silnej dodatniej zależności parametrów. W
praktyce oznacza to, iż wraz ze wzrostem wartości jednego parametru obserwuje się
wzrost wartości drugiego. Jeśli współczynnik korelacji jest bliski -1 to parametry są
ujemnie skorelowane. Wartość współczynnika korelacji bliska zeru świadczy o
niezależności dwóch parametrów. Ich wzajemne podobieństwa możemy przedstawić
w postaci kwadratowej i symetrycznej macierzy kowariancji (cov(xk,xl) = cov(xl,xk))
lub macierzy współczynników korelacji (r(xk,xl) = r(xl,xk)), które mają wymiary n×n.
Na Rys. 4 przedstawiono macierz współczynników korelacji uzyskanych dla
parametrów macierzy danych z Rys. 2a. Ma ona wymiary 2×2. Elementy jej
przekątnej są równe jeden, gdyż pomiędzy dwiema tymi samymi zmiennymi istnieje
idealna korelacja. Wartość współczynnika korelacji i jego znak świadczą o silnej
zależności tych parametrów jak i dodatnim charakterze tej zależności.
0,9628
0,9628
1
indeks parametru
1
indeks parametru
Rys. 4 Macierz współczynników korelacji uzyskana dla parametrów macierzy X,
którą przedstawiono na Rys. 2a.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
6
Na Rys. 5 schematycznie przedstawiono dodatnią i ujemną korelację oraz jej brak dla
dwóch symulowanych parametrów.
c)
a) 13
17.5
17
16.5
12
16
zmienna 2
zmienna 2
11
10
9
15.5
15
14.5
14
8
13.5
13
7
5.5
6
6.5
7
7.5
8
8.5
9
9.5
10
12.5
1.5
zmienna 1
b)
2
2.5
3
3.5
4
4.5
5
zmienna 1
8
7
zmienna 2
6
5
4
3
2
5.5
6
6.5
7
7.5
8
8.5
9
9.5
10
zmienna 1
Rys. 5 Projekcja 40 próbek na przestrzeń dwóch symulowanych parametrów, które są: a) skorelowane
dodatnio (r = 0,8309), b) skorelowane ujemnie (r = -0,8309) i c) praktycznie nieskorelowane
(r = 0,0705).
Zależność pomiędzy parametrami, które tworzą wielowymiarowe dane chemiczne jest
kluczową własnością i dzięki niej możliwa jest redukcja ich wymiarowości, a co za
tym idzie możliwa jest ich wizualizacja. Parametry, które są zależne da się zastąpić
kilkoma nowymi zmiennymi, które są liniowymi kombinacjami oryginalnych
parametrów, bez utraty istotnej chemicznie informacji. Zmienna, silnie skorelowana z
innymi, nie wnosi dodatkowej informacji o zróżnicowaniu danych, gdyż informacja,
jaką opisuje jest już zawarta w innych zmiennych. Ilość tych nowych zmiennych,
która wystarcza do opisu badanego układu lub zjawiska mówi o jego kompleksowości.
W przypadku dużej liczby parametrów bezpośrednia wizualizacja wielowymiarowych
danych jest niemożliwa. Do ich wizualizacji stosuje się różnego rodzaju techniki
eksploracji danych. Mają one na celu ułatwić odpowiedzi na takie pytania jak:
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
7
•
Które próbki są do siebie podobne w przestrzeni mierzonych parametrów?
•
Które z mierzonych parametrów zawierają podobną informację o badanych
próbkach (są zależne)?
•
Które z parametrów mają największy wkład do obserwowanych podobieństw
(czy też różnic) pomiędzy próbkami?
•
Jaka jest kompleksowość badanego układu lub zjawiska?
Ogólnie wyróżniamy dwie główne grupy technik eksploracji wielowymiarowych
danych:
•
metody projekcji [3], wśród których analiza czynników głównych (z ang.
principal component analysis, PCA) [4] ma swoje szczególne miejsce, oraz
•
metody grupowania danych [5,6].
W wielu metodach projekcyjnych, redukcja wymiarowości danych oparta jest o
konstrukcję nowych zmiennych, które są liniową kombinacją oryginalnych
zmiennych. Jedną z metod projekcyjnych jest metoda poszukiwania projekcji (z ang.
projection pursuit) [7]. To najbardziej uniwersalna metoda tego typu, ponieważ w
zależności od użytego do poszukiwania projekcji kryterium, pozwala otrzymać
rozwiązania innych technik projekcji [8,9,10]. W metodzie poszukiwania projekcji
konstruuje się w wielowymiarowej przestrzeni danych kierunki, które mają na celu
ujawnić „ciekawą” strukturę danych. W rzeczywistości, to czy dany kierunek i
odpowiadająca mu projekcja jest „ciekawa” określa tzw. indeks projekcji [11]. Wśród
wielu możliwych indeksów projekcji znajdziemy wariancję, czy indeksy takie jak
entropia [8] lub kurtoza [12,13], opisujące na ile rozkład projekcji różni się od
rozkładu normalnego. Projekcje o rozkładzie normalnym są uznawane za najmniej
interesujące. Zależnie od użytego indeksu projekcji, metoda poszukiwania projekcji
może prowadzić np. do konstrukcji czynników głównych (maksymalizacja wariancji
projekcji), stabilnych czynników głównych (maksymalizacja stabilnej skali projekcji
[14]) albo ukrytych zmiennych, które ujawniają grupy w danych (np. stosując jako
indeks projekcji kurtozę [10]). Na Rys. 6, dla symulowanych dwuwymiarowych
danych, zawierających dwie grupy obiektów (po 40 każda), przedstawiono dwa
„ciekawe” kierunki i odpowiadające im projekcje, skonstruowane na podstawie
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
8
indeksów wariancji oraz entropii. Jak widać na Rys. 6b-e wybór indeksu projekcji
pozwala na ujawnienie na projekcjach różnych aspektów struktury danych (np.
podgrup obiektów czy obiektów odległych). W przypadku maksymalizacji wariancji
projekcji, projekcja jest tak konstruowana, aby opisywała najlepiej wariancję danych.
a)
5
4
kierunek 2
kierunek 1
3
zmienna 2
2
1
0
-1
-2
-3
-4
-2
-1.5
-1
-0.5
0
0.5
1
1.5
zmienna 1
c)
12
4
10
ilość obiektów
5
współrzędna obiektu na kierunku 1
b)
8
6
4
2
3
2
1
0
-1
-2
-3
-4
0
-5
-4
-3
-2
-1
0
1
2
3
4
-5
5
0
10
20
d)
e)
18
współrzędna obiektu na kierunku 2
14
12
ilość obiektów
40
50
60
70
80
60
70
80
2.5
2
16
10
8
6
4
2
0
30
indeks obiektu
współrzędne obiektów na kierunku 1
1.5
1
0.5
0
-0.5
-1
-1.5
-2
-2.5
-2
-1.5
-1
-0.5
0
0.5
1
1.5
2
2.5
-2.5
0
współrzędne obiektów na kierunku 2
10
20
30
40
50
indeks obiektu
Rys. 6 a) Dwa kierunki poprowadzone w dwuwymiarowej przestrzeni danych, b) histogram
współrzędnych obiektów uzyskanych po ich ortogonalnej projekcji na pierwszy kierunek [0,7071 0]
oraz c) współrzędne obiektów tej projekcji, d) histogram współrzędnych obiektów uzyskanych po ich
ortogonalnej projekcji na drugi kierunek [0 -0,7071] i e) wartości współrzędnych obiektów na tej
projekcji.
Dla rozważanych danych projekcja obiektów na pierwszy kierunek nie ujawnia grup
w danych, gdyż te nie są rozmieszczone wzdłuż osi o największej wariancji, a
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
9
histogram tejże projekcji ma rozkład zbliżony do rozkładu normalnego (zob. Rys. 6b i
c). Maksymalizując entropię projekcji, kładzie się nacisk na uchwycenie projekcji o
rozkładzie dalekim od normalnego. W tym przypadku histogram projekcji ma rozkład
bimodalny, co świadczy o obecności w danych dwóch grup (zob. Rys. 6d i e). Nie
wszystkie indeksy projekcji mogą prowadzić do ujawnienia na projekcjach grup, jak
np. dla projekcji danych prezentowanej na Rys. 6c.
2. ANALIZA CZYNNIKÓW GŁÓWNYCH
Analiza czynników głównych, jest szczególnym przypadkiem metody poszukiwania
projekcji, w której jako indeks projekcji używa się wariancję. PCA stosuje się
głównie do modelowania, kompresji i wizualizacji wielowymiarowych danych
[4,15,16,17]. Za pioniera PCA uważa się Pearsona. W 1901 roku opublikował on
pracę o prostych i płaszczyznach, które są najlepiej dopasowane do zbioru próbek w
przestrzeni pomiarowej [ 18 ]. Następnie, po 22 latach Fisher i MacKenzie [ 19 ]
zaproponowali pierwszy algorytm do PCA, znany obecnie jako algorytm NIPALS,
który ponownie odkrył Wold w 1966 roku [20]. Kolejne modyfikacje metody PCA
zawdzięczamy Hottelingowi [21].
Zadaniem PCA jest przedstawienie danych, X, o m obiektach i n zmiennych, jako
iloczyn dwóch nowych macierzy T (m×f) i P (n×f), gdzie f<<n, które zawierają
współrzędne obiektów i parametrów na kierunkach maksymalizujących opis wariancji
danych. O liczbie kolumn macierzy T i P, czyli o kompleksowości modelu PCA,
decyduje rząd chemiczny macierzy X, który jest co najwyżej równy jej rzędowi
matematycznemu, o czym szerzej napiszemy w dalszej części rozdziału. Model PCA
można wyrazić następująco:
X [ m ,n ] = T[ m , f ] P[Tf ,n ] + E [ m ,n ]
(5)
gdzie E to macierz reszt od modelu PCA z f czynnikami głównymi.
Kolumny macierzy T i P zawierają współrzędne obiektów i parametrów na nowych
ukrytych zmiennych, nazywanych czynnikami głównymi. Czynniki główne są
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
10
konstruowane iteracyjnie tak, aby maksymalizować opis wariancji danych. Każdy
kolejny czynnik główny opisuje niewyjaśnioną przez poprzednie czynniki wariancję
danych i dlatego jego wkład do opisu całkowitej wariancji danych jest mniejszy.
Kolumny T są ortogonalne, a kolumny P ortonormalane - czyli mają jednostkową
długość i są ortogonalne. Biorąc pod uwagę wspomniane ograniczenia dotyczące
konstrukcji czynników głównych równanie 5 ma rozwiązanie.
Z każdym czynnikiem głównym jest stowarzyszona, tzw. wartość własna, vi. Oblicza
się ją jako sumę kwadratów wartości wyników dla danego czynnika głównego.
Wartości własne określają ilościowo wariancję danych opisaną przez kolejne czynniki.
Procent całkowitej wariancji danych, I, jaką opisuje f kolejnych czynników głównych
można obliczyć jako:
f
I=
∑v
i =1
m n
i
∑∑ xij2
⋅ 100
(6)
i =1 j =1
gdzie, xij to poszczególne elementy centrowanej macierzy X.
Schematycznie, model PCA przedstawiono na Rys. 7.
p1
m
p2
+
=
t1
X[m,n]
n
pf
+
+ ,,, +
m
m
m
m
Dane wyjściowe
n
n
n
n
t2
tf
Macierz reszt
E[m,n]
n
+
=
+
+ ,,, +
m
m
Dane wyjściowe
X[m,n]
X[m,n](1)
X[m,n](2)
X[m,n](f)
Macierz reszt
E[m,n]
dane wyjściowe zrekonstruowane z 1, 2, …, f czynnikami głównymi
Rys. 7 Model PCA o f czynnikach głównych.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
11
Model PCA ma stosunkowo prostą interpretację graficzną. Na Rys. 8a przedstawiono
symulowany dwuwymiarowy zbiór danych, który zawiera sześć obiektów,
oznaczonych jako ({). Naszym zadaniem będzie skonstruowanie dwóch czynników
głównych, mając na uwadze, że powinny one jak najlepiej opisać wariancję danych.
Na Rys. 8a przedstawiono kierunek i odpowiadającą mu jednowymiarową projekcję,
która maksymalizuje opis wariancji danych.
b)
a)
PC 2
PC 1
1
+
0.5
zmienna 2
zmienna 2
0.5
++
0
-0.5
-1
PC 1
1
+
-1
++
-0.5
+
+
+
+
+
0
-0.5
0
zmienna 1
0.5
1
1.5
-1
-1
-0.5
0
0.5
1
1.5
zmienna 1
Rys. 8 Projekcje obiektów ({) na płaszczyzny zdefiniowane przez zmienne 1 i 2: a) kierunek, który
maksymalizuje wariancję projekcji i projekcja (PC 1) z zaznaczonymi wartościami wyników (+), oraz
b) pierwsze dwa kierunki i projekcja obiektów (PC 2) na drugi kierunek z zaznaczonymi wartościami
wyników (+) (PC 2 opisuje część informacji, jakiej nie modeluje pierwszy czynnik główny).
Jeśli poprzestaniemy na modelu PCA tylko z jednym czynnikiem głównym, wówczas
odległości prowadzone od każdego punktu ({) prostopadle do PC 1, jak pokazano na
Rys. 8a, będą odpowiadały resztom od modelu. Biorąc pod uwagę tylko pierwszą
projekcję, wymiarowość danych zostanie zredukowana z dwóch wymiarów do
jednego. Aby opisać pozostałą część wariancji danych, drugi kierunek musi być
prostopadły do pierwszego (zob. Rys. 8b). Jeśli dane mają więcej niż dwa wymiary,
wówczas następne kierunki są prostopadłe do tych już wytyczonych.
Czynniki główne tworzą nowy układ współrzędnych, w którym odległości
euklidesowe pomiędzy obiektami są zachowane (tzn. są równe odległościom w
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
12
przestrzeni oryginalnych zmiennych). Każdy obiekt ma współrzędne określone przez
odpowiednie wyniki, [ti1, ti2, ..., tif].
Do konstrukcji czynników głównych można stosować różne algorytmy [22], a wśród
nich są takie jak np. NIPALS, SVD, EVD, [23]. Na szczególną uwagę zasługują
szybkie algorytmy PCA, w których czynniki główne powstają poprzez dekompozycję
kwadratowej macierzy XXT albo XTX [24]. Kwadratowa macierz tworzona jest tak,
aby jej wymiar był najmniejszy, co zapewnia szybką konstrukcję czynników
głównych. Macierz XTX dla danych centrowanych nazywana jest macierzą wariancjikowariancji [ 25 ]. Elementy diagonali tej macierzy to wariancje poszczególnych
parametrów, a pozostałe elementy wyrażają ich kowariancje. Jeśli dane poddano
autoskalowaniu, wówczas macierz XTX jest macierzą korelacji, a jej elementy to
współczynniki korelacji Pearsona [2].
2.1 Wstępne przygotowanie danych do dalszej analizy
Wstępne przygotowanie danych ma na celu (i) podnieść ich jakość oraz (ii) poprawić
interpretację danych. Istnieje wiele metod wstępnego przygotowania danych [23].
Metody przygotowania danych do dalszej analizy możemy podzielić na trzy grupy.
Pierwsza z nich obejmuje metody stosowane do indywidualnych obiektów macierzy
danych, np. sygnałów instrumentalnych (metody eliminacji szumu i linii bazowej,
różnego rodzaju procedury normalizacyjne, pochodne, itp.).
Do drugiej grupy metod zaliczamy techniki, których zadaniem jest modyfikacja
indywidualnych zmiennych (metody centrowania i/lub skalowania indywidualnych
zmiennych, np. standardyzacja, autoskalowanie i transformacja logarytmiczna), a
także metody eliminacji zmiennych, które mogą być uznawane jako skrajny wariant
modyfikacji zbioru zmiennych [26,27]).
Trzecia grupa metod to metody stosowane do nakładania sygnałów instrumentalnych.
Poniżej przedstawiono najczęściej stosowane metody wstępnego przygotowania
danych.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
13
2.1.1 Centrowanie i skalowanie danych
Najczęściej stosowaną transformacją danych jest centrowanie. Ma ona na celu usunąć
z danych stałe elementy, które nic nie wnoszą do wiedzy o zróżnicowaniu danych.
Centrowanie polega na odjęciu od każdego elementu kolumny odpowiedniej wartości
średniej.
Kolejną możliwą operacją jest autoskalowanie. Stosuje się je, gdy parametry
zmierzono w różnych jednostkach i/lub ich zakresy zmienności znacznie się różnią.
Autoskalowanie polega na centrowaniu kolumn danych, a następnie podzieleniu
każdego elementu określonej kolumny przez jej odchylenie standardowe. Wynikiem
takiej operacji jest nadanie każdej zmiennej jednostkowej wariancji, a więc tej samej
wagi w późniejszej analizie. Na Rys. 9 przedstawiono średnie i odchylenia
standardowe zmiennych przed i po autoskalowaniu dla symulowanych danych
zawierających sto próbek i dwadzieścia parametrów.
c)
100
10
80
9
70
60
średnia
11
90
odchylenie standardowe
a)
50
40
30
20
7
6
5
4
3
2
10
0
8
1
0
2
4
6
8
10
12
14
16
18
0
20
0
2
4
6
indeks parametru
b)
8
10
12
14
16
18
20
14
16
18
20
indeks parametru
d)
1
1
0.8
odchylenie standardowe
0.6
0.4
średnia
0.2
0
-0.2
-0.4
-0.6
0.8
0.6
0.4
0.2
-0.8
-1
0
2
4
6
8
10
12
indeks parametru
14
16
18
20
0
0
2
4
6
8
10
12
indeks parametru
Rys. 9 Wartości średnie dwudziestu parametrów symulowanych danych a) przed i b) po operacji
autoskalowania oraz odpowiadające im odchylenia standardowe c) przed i b) po autoskalowaniu.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
14
Podkreślmy jeszcze raz, iż PCA prowadzona dla autoskalowanych parametrów
oznacza, iż czynniki główne otrzymuje się w oparciu o macierz korelacji. Typowym
przykładem danych, jakie zazwyczaj wymagają takiego właśnie przygotowania, są
dane środowiskowe, gdyż tworzą je parametry fizyko-chemiczne mierzone w różnych
jednostkach i zakresach. W literaturze, autoskalowanie nazywane jest także ztransformacją lub skalowaniem zmiennych do jednostkowej wariancji [23].
W przypadku danych, w których wyróżnia się bloki zmiennych, np. blok widm
Ramana i blok widm UV-VIS, skalowaniu można poddać indywidualne bloki
zmiennych tak, by wariancja każdego z nich była równa jedności [4,28].
Dla uzyskania bardziej symetrycznych rozkładów zmiennych, przypominających
rozkład normalny, często stosuje się transformację logarytmiczną. Zazwyczaj, takiej
transformacji wymagają dane zawierające informacje o elementach śladowych i
niejednokrotnie dane środowiskowe [4].
2.1.2 Normalizacja sygnałów
Normalizacja indywidualnych sygnałów macierzy danych ma na celu usunięcie efektu
związanego z różną ilością próbki użytej w eksperymencie (np. w chromatografii różna objętość wprowadzonej na kolumnę próbki). Normalizacja polega na
podzieleniu każdego elementu wiersza macierzy przez jego długość (tj. pierwiastek
sumy kwadratów wszystkich elementów danego wiersza macierzy). W wyniku
normalizacji długość każdego wektora jest jednostkowa.
Innym typem normalizacji jest transformacja SNV (z ang. standard normal variate)
[29], często stosowana np. do korekcji widm z bliskiej podczerwieni. Jej celem jest
transformacja poszczególnych sygnałów tak, aby ich wariancja była jednostkowa. W
tym celu wiersze macierzy centruje się odpowiadającymi im wartościami średnimi, a
następnie dzieli się przez ich odchylenia standardowe. Na Rys. 10 przedstawiono
zbiór widm z bliskiej podczerwieni przed i po transformacji SNV.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
15
a)
b)
2
1.2
SNV-transformowana absorbancja
1.5
1.1
absorbancja
1
0.9
0.8
0.7
0.6
0.5
0.4
1
0.5
0
-0.5
-1
-1.5
-2
0.3
1200
1400
1600
1800
2000
2200
2400
1200
długość fali [nm]
1400
1600
1800
2000
2200
2400
długość fali [mn]
Rys. 10 Zbiór stu widm z bliskiej podczerwieni próbek zboża a) przed i b) po transformacji SNV.
Wybór odpowiedniej metody wstępnego przygotowania danych nie jest oczywisty i
wywiera wpływ na konstrukcję czynników głównych oraz na ich późniejszą
interpretację, co zademonstrowano w paragrafie 4.3.
2.2 Efektywność kompresji danych
Zastanówmy się teraz, kiedy kompresja danych do kilku czynników głównych będzie
skuteczna. Zgodnie z regułami algebry liniowej, dla macierzy X można skonstruować
fmax czynników głównych, gdzie fmax to matematyczny rząd macierzy danych. Rząd
macierzy to maksymalna liczba wektorów bazowych, które wystarczają w zupełności
do jej opisu [30]. Rząd macierzy może być równy, co najwyżej, minimum z jej dwóch
wymiarów, min(n,m). Tak więc, w zależności od wymiarowości danych, macierz
danych może mieć maksymalny rząd równy liczbie obiektów lub zmiennych. Dla
centrowanej macierzy danych, gdzie m<n, jej maksymalny rząd wynosi m-1, a dla
centrowanej macierzy o większej liczbie wierszy niż kolumn, jej maksymalny rząd
wynosi n [23].
W praktyce, faktyczny rząd macierzy, nazwijmy go rzędem chemicznym, jest często
dużo mniejszy niż jej rząd matematyczny. Spowodowane jest to tym, iż wszystkie
dane pomiarowe obarczone są błędem eksperymentalnym, a zatem tylko kilka
pierwszych czynników głównych, o dużych wartościach własnych modeluje dane, a
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
16
pozostałe modelują jedynie błąd eksperymentalny. Kompresja danych do kilku
czynników głównych jest tym skuteczniejsza im więcej jest w danych silnie
skorelowanych zmiennych.
2.3 Wybór kompleksowości modelu PCA
W zależności od zastosowań PCA, wybór liczby czynników głównych do modelu
PCA może mieć różne znaczenie. W przypadku użycia PCA do eksploracji danych,
zazwyczaj skupiamy się na interpretacji projekcji obiektów i zmiennych na kilka
pierwszych czynników głównych, gdyż właśnie one modelują przeważającą wariancję
danych. Wówczas ustalenie liczby czynników głównych nie jest krytyczne.
Inaczej jest, gdy metoda PCA jest użyta do kompresji danych, a macierz wyników ma
zastąpić oryginalne dane. Wtedy, do modelu PCA należy wybrać optymalną liczbę
czynników głównych. Wybór optymalnej liczby czynników do modelu PCA jest
bardzo ważny, gdyż pozwala na eliminację części błędu eksperymentalnego z danych,
a jednocześnie zapewnia, że nie nastąpi utrata istotnej chemicznie informacji. Istnieje
wiele sposobów ułatwiających wybór optymalnej liczby czynników głównych, np.
analiza wartości własnych lub wariancji, jaką opisują kolejne czynniki główne. Inne
metody bazują na różnych indeksach, np. indeksie Malinowskiego [30] lub [31,32].
Kolejnym sposobem jest metoda kroswalidacji, zwana także walidacją krzyżową. Ma
ona wiele wariantów, a wyczerpujący przegląd technik kroswalidacji czytelnik
znajdzie w [33]. Najpopularniejszym typem kroswalidacji jest kroswalidacja typu
„wyrzuć jeden obiekt”. W metodzie tej konstruuje się m modeli PCA o rosnącej
liczbie czynników głównych. Modele te budowane są dla podzbiorów danych
powstałych poprzez usuwanie z wyjściowych danych kolejno każdego obiektu.
Usunięty obiekt to tzw. obiekt testowy i służy on do oceny mocy predykcyjnej modeli
PCA o różnej liczbie czynników głównych na podstawie reszt od modelu dla tego
obiektu. Reszty oblicza się jako różnice pomiędzy wartościami parametrów dla i-tego
obiektu, a wartościami zrekonstruowanymi stosując model o f czynnikach głównych,
gdzie f = 1, 2, ..., fmax:
t [1, f ] = x [1,n ] P[ n , f ]
(8)
e[1,n ] ( f ) = x [1,n ] − t [1, f ] P[Tf ,n ]
(9)
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
17
Dla każdego obiektu testowego, sumuje się uzyskane kwadraty reszt uzyskane od
modeli z 1, 2,..., fmax czynnikami głównymi (zob. równanie 9), otrzymując wektor
kwadratów reszt, o wymiarze (1×fmax). Następnie, te wektory zestawia się macierz
CVE.
Po zakończeniu procedury kroswalidacji „wyrzuć jeden obiekt”, macierz CVE ma
wymiary (m×fmax). Na jej podstawie oblicza się średni błąd kwadratowy kroswalidacji,
RMSECV, zgodnie z wzorem:
RMSECV =
1 m
⋅ ∑ ( CV eij )
m i =1
(10)
Idee procedury kroswalidacji typu „wyrzuć jeden obiekt” obrazuje Schemat 1. Z
teoretycznego punktu widzenia, optymalna liczba czynników głównych w modelu
PCA zapewnia możliwie najmniejszy błąd przewidywania modelu.
n
Usuwanie w
kolejnych krokach
i-tego obiektu z
macierzy
Konstrukcja modeli
PCA o coraz
większej liczbie
czynników głównych
m-1
f
m
RMSECV =
1
⋅∑
m i =1
Obliczenie kwadratów reszt
od modelu dla i-tego
obiektu w oparciu o modele
z różną liczbą czynników
głównych (1, 2, ..., f)
CVE
m
Obliczenie średniego błędu
kwadratowego
kroswalidacji na podstawie
kwadratów reszt od modeli
PCA dla każdego i-tego
obiektu
Schemat 1. Przedstawienie idei kroswalidacji typu „wyrzuć jeden obiekt”.
Dla dużej liczby czynników głównych model dobrze rekonstruuje dane, ale jego
przewidywanie dla nowych próbek jest złe. Stąd wykres wartości RMSECV od liczby
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
18
czynników powinien charakteryzować się minimum, które wskazuje optymalną
kompleksowość modelu PCA. W praktyce, ze względu na szum w danych, wykresy
RMSECV nie zawsze mają wyraźne minimum, a przez to wybór optymalnej liczby
czynników nie jest oczywisty.
Najczęściej spotykane typy krzywych RMSECV zaprezentowano na Rys. 11. Jedynie
krzywa oznaczona jako (−{−) pozwala pewnie stwierdzić, iż model PCA powinien
zawierać cztery czynniki główne.
11
10
9
8
RMSECV
7
6
5
4
3
2
1
0
0
1
2
3
4
5
6
7
8
9
10
liczba czynników głównych
Rys. 11 Przykładowe krzywe błędu kroswalidacji (RMSECV) w zależności od liczby
czynników głównych w modelu PCA - z wyraźnym minimum (−{−) i bez (−−).
W porównaniu z krzywą (−{−), na podstawie krzywej (−−) wybór optymalnej
liczby czynników do modelu jest znacznie trudniejszy. Analizując zmiany kolejnych
wartości RMSECV dla modeli z f i f+1 czynnikami można stwierdzić, że model PCA
najprawdopodobniej powinien mieć cztery lub pięć czynników głównych. W
trudniejszych przypadkach należy się posiłkować innymi niż kroswalidacja metodami
wyboru czynników głównych [30].
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
19
3. RÓŻNE ZASTOSOWANIA METODY PCA I JEJ MODYFIKACJE
W literaturze można zaleźć szereg atrakcyjnych zastosowań PCA. Najczęściej metoda
ta jest wykorzystywana jako technika wizualizacji danych. Obecnie, rutynowa analiza
wielowymiarowych danych zakłada ich wstępną eksplorację, co zazwyczaj skutecznie
umożliwia metoda PCA.
Oprócz typowych zastosowań eksploracyjnych, PCA używa się także do kompresji
danych, zastępując oryginalne zmienne kilkoma czynnikami głównymi, które opisują
przeważającą część wariancji danych. Stąd PCA jest również traktowana jako etap
wstępnego przygotowania danych do dalszej analizy, a zabieg kompresji danych ma
na celu przyspieszenie lub uproszczenie kolejnych obliczeń. Przykłady takiego użycia
PCA znajdujemy w modelowaniu danych sieciami neuronowymi [ 34 ], podczas
grupowania danych [35], konstrukcji stabilnych czynników głównych [15] czy też w
niektórych metodach regresji, np. [36,37]. Istnieją także metody, które wymagają
nieskorelowanych zmiennych. Prostym sposobem na pozbycie się skorelowanych
zmiennych jest zastąpienie ich czynnikami głównymi. Ma to miejsce, np. w
przypadku obliczania odległości Mahalanobisa [38], regresji czynników głównych
[36,37] oraz w technice minimalnego wyznacznika kowariancji [15].
Odpowiednie stosowanie metody PCA pozwala również na badanie czystości
mieszanin, np. poprzez analizę sygnałów instrumentalnych otrzymanych sprzężonymi
technikami chromatograficznymi [39].
3.1. Eksploracja danych zawierających obiekty odległe i/lub brakujące elementy
PCA jest bardzo ogólną techniką modelowania danych. Jednakże, w niektórych
przypadkach wymaga ona pewnych modyfikacji. Ma to miejsce w szczególności, gdy
obiektem analizy są dane z obiektami odległymi (czyli z próbkami bardzo różniącymi
się od pozostałych) i/lub brakującymi elementami.
Poniżej omówiono stabilną metodę PCA, która pozwala na analizę danych z
obiektami odległymi oraz modyfikację metody PCA stosowaną do analizy danych z
brakującymi elementami.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
20
3.1.1 Stabilny wariant PCA
Jak już wspomniano, zadaniem PCA jest maksymalizować opis wariancji danych.
Zatem, poszukuje się takich kierunków, aby projekcja obiektów na te kierunki
charakteryzowała się maksymalną wariancją. W związku z tym, obiekty odległe
wpływają na konstruowane czynniki główne, a model PCA opisuje głównie te obiekty
[40,41].
Do tej pory w literaturze zaproponowano wiele algorytmów do konstrukcji tak
zwanych stabilnych czynników głównych, na których konstrukcję nie wpływają
obiekty odległe [42,43,44,45]. W tym rozdziale przedstawimy metodę Crouxa i RuizGazena [14], ze względu na jej dużą prostotę w porównaniu z innymi stabilnymi
wariantami PCA. W metodzie Crouxa i Ruiz-Gazena zamiast maksymalizować
wariancję projekcji, szuka się projekcji o największej wartości stabilnego estymatora
skali, tzw. estymatora Qn [46]. Każdy stabilny estymator ma za zadanie poprawnie
estymować określoną własność (np. średnią czy odchylenie standardowe) nawet, jeśli
w danych występują obiekty odległe. Koncepcje stabilnych estymatorów oraz
stabilnych metod zostały omówione w [47,48,49].
Kolejne kroki tworzenia stabilnych czynników głównych w oparciu o algorytm
Crouxa i Ruiz-Gazena można przedstawić następująco:
1. centrowanie danych stosując stabilny estymator średniej (medianę lub
L1-medianę [50]),
2. konstrukcja m kierunków, będących znormalizowanymi wierszami
macierzy danych,
3. projekcja m obiektów na m kierunków,
4. znalezienie takiego kierunku, dla którego projekcja obiektów na ten
kierunek osiąga największą wartość stabilnego estymatora skali Qn,
5. usunięcie z macierzy danych informacji opisanej przez i-ty stabilny czynnik
główny,
6. powrót do kroku 2, jeśli konieczna jest konstrukcja dodatkowych stabilnych
czynników głównych.
Ponieważ stabilny model PCA nie jest zaburzony obiektami odległymi, dlatego
umożliwia on ich diagnostykę. Przeprowadza się ją zazwyczaj w oparciu o reszty od
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
21
stabilnego modelu PCA i odległości Mahalanobisa obliczone w przestrzeni f
stabilnych czynników głównych [44]. Odległość Mahalanobisa [38] dla danych
uprzednio centrowanych wyraża odległość i-tego obiektu od środka danych w
przestrzeni stabilnego modelu:
di =
∑ (t
f
j =1
/vj )
2
ij
(11)
gdzie, ti to stabilne wartości f wyników dla i-tego obiektu, a vj to stabilne wartości
własne j-tego czynnika głównego.
Aby ułatwić diagnostykę obiektów odległych, zarówno wektor reszt od stabilnego
modelu (pierwiastek sumy kwadratów reszt obliczonych zgodnie z równaniem 9) jak i
wektor odległości Mahalanobisa każdego obiektu poddaje się z-transformacji.
Z-transformowane elementy wektora to absolutne wartości różnic pomiędzy każdym
elementem wektora (reszt i odległości Mahalanobisa), a jego medianą, podzielone
następnie przez odchylenie standardowe wektora, oszacowane stabilnym estymatorem
skali, np. estymatorem Qn [49]:
dZ i =
d i − med (d )
Qn(d )
(12)
gdzie, dZi to z-transformowana wartość i-tej wartości wektora reszt lub odległości
Mahalanobisa, ‘med(d)’ oznacza medianę wektora d, a ‘Qn(d)’ to estymowana
wartość odchylenia standardowego wektora d stosując stabilny estymator skali Qn.
Taki zabieg pozwala na łatwe wyznaczenie wartości progowych dla ztransformowanych reszt i odległości Mahalanobisa, zakładając, że ich rozkład jest
normalny. Wtedy, dla 99,9% obiektów wartości z-transformowanych reszt i
odległości Mahalanobisa będą poniżej wartości progowej równej trzy. Ze względu na
wartości z-transformowanych reszt i odległości Mahalanobisa każdy obiekt można
przypisać do jednej z czterech kategorii (zob. Rys. 12).
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
22
reszty od modelu
*
*
2 4
* *
*
** * *
*
***
*
* *
* *
1 3
*
odległość Mahalanobisa
Rys. 12 Diagram reszt od stabilnego modelu PCA i odległości Mahalanobisa,
obliczonych w przestrzeni f stabilnych czynników głównych.
Mianowicie, wyróżniamy obiekty:
1. regularne, czyli te o małych wartościach reszt od stabilnego modelu i małych
odległościach Mahalanobisa,
2. o dużych resztach od modelu, przekraczających wartość progową,
3. o wartościach odległości Mahalanobisa powyżej wartości progowej, oraz
4. obiekty o wartościach reszt od modelu i odległości Mahalanobisa większych
od wartości progowych.
Obiekty czwartej kategorii mają największy wpływ na konstrukcję czynników
głównych.
3.1.2 PCA dla danych z brakującymi elementami
Metoda PCA do analizy danych z brakującymi elementami, EM-PCA, bazuje na
procedurze maksymalizacji wartości oczekiwanych (z ang. expectation-maximization
principal component analysis). Na początku, brakujące elementy zastępuje się
wartościami oczekiwanymi (średnimi ze średnich wartości kolumn i wierszy macierzy
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
23
danych). Następnie, iteracyjnie estymuje się brakujące elementy stosując model PCA
z f czynnikami głównymi. Brakujące elementy estymuje się do momentu, gdy suma
kwadratów różnic pomiędzy estymowanymi wartościami brakujących elementów w
dwóch kolejnych iteracjach jest znikomo mała [ 51]. Kryterium zbieżności, S, w
metodzie EM-PCA wyraża się jako:
S = ∑∑ (eijk +1 − eijk ) 2
i
(13)
j
gdzie, S to suma kwadratów różnic reszt pomiędzy estymowanymi elementami
macierzy o indeksach ij, których nie było w wyjściowej macierzy danych, uzyskane w
k-tej ( eijk ) i k+1 iteracji ( eijk +1 ).
Estymacja brakujących elementów jest skuteczna, jeśli w danych istnieje stosunkowo
dobra struktura korelacyjna, a brakujące elementy są estymowane tak, aby nie
zaburzać końcowego modelu PCA.
Kolejne kroki algorytmu EM-PCA, w którym brakujące elementy estymowane są
stosując model z f czynnikami głównymi, można przedstawić następująco (zob.
Schemat 2):
1. ustalenie wartości progowej kryterium zbieżności (np. S = 10-4) i wstępne
podstawienie brakujących elementów macierzy danych ich wartościami
oczekiwanymi (tzn. średnimi ze średnich wartości kolumn i średnich wartości
wierszy macierzy),
2. przygotowanie danych (np. centrowanie lub autoskalowanie dla
obserwowanych elementów),
3. dekompozycja macierzy danych do f czynników głównych,
4. rekonstrukcja danych stosując model PCA o f czynnikach głównych,
5. podstawienie brakujących elementów w macierzy danych estymowanymi
wartościami w kroku 4 algorytmu,
6. sprawdzenie kryterium zbieżności algorytmu (równanie 13), a jeśli to
konieczne powrót do kroku 2.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
24
Optymalną liczbę czynników w modelu EM-PCA ustala się stosując np.
kroswalidację.
n
Wstępne
podstawienie
brakujących
elementów
X[ m ,n ] = T[ m , f ] P[Tf ,n ] + E[ m ,n ]
m
Konstrukcja modelu PCA
z f czynnikami głównymi
Dane wyjściowe
X[m,n]
NIE
Konwergencja?
Podstawienie brakujących
elementów wartościami
przewidzianymi na
podstawie modelu PCA
o f czynnikach głównych
TAK
Konstrukcja nowego modelu
PCA z f czynnikami głównymi
Dane z podstawionymi
brakującymi elementami
Schemat 2. Główne kroki iteracyjnej procedury EM-PCA.
4. KONSTRUKCJA MODELI PCA
Do tej pory, zwróciliśmy uwagę na kilka ważnych własności PCA, a mianowicie:
1. PCA jest modelem, który aproksymuje dane. Jakość aproksymacji zależy od ilości
czynników głównych użytych do konstrukcji modelu. Uwzględnienie optymalnej
liczby czynników głównych w modelu pozwala na częściową eliminację szumu z
danych eksperymentalnych,
2. czynniki główne są nowymi ortogonalnymi zmiennymi (wyrażane są jako liniowa
kombinacja oryginalnych zmiennych) i maksymalizują opis wariancji danych,
3. czynniki główne tworzą nowy układ współrzędnych [52],
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
25
4. stosując metodę PCA, macierz danych jest przedstawiana jako iloczyn dwóch
nowych macierzy, macierzy wyników, T, i wag, P. Zawierają one odpowiednio
informacje o obiektach i zmiennych eksperymentalnych,
5. na konstrukcję czynników głównych istotny wpływ wywierają obiekty odległe.
W tej części rozdziału skupimy się na zilustrowaniu w/w własności PCA oraz
zaprezentujemy niektóre zastosowania PCA do analizy eksperymentalnych danych.
4.1 Opis danych eksperymentalnych, jakich użyto do dyskusji
Praktyczne zalety metody PCA zademonstrujemy w oparciu o cztery zestawy danych,
których wybór był podyktowany ich ogólną dostępnością.
Dane 1 tworzy 100 widm próbek zboża, które zarejestrowano stosując technikę
spektroskopii w bliskiej podczerwieni (NIR) [53]. Widma odbiciowe zmierzono w
zakresie spektralnym 1100 - 2500 nm, co 2 nm. Dane są dostępne z [54].
Dane 2 to wyniki analiz 178 próbek włoskich win takich jak Barolo (59 próbek),
Grignolino (71 próbek) i Barbera (48 próbek). Dla każdej próbki oznaczono
trzynaście parametrów (1- zawartość alkoholu, 2- zawartość kwasu jabłkowego, 3ilość popiołu, 4- zasadowość popiołu, 5- zawartość magnezu, 6- całkowita zawartość
fenoli,
7- zawartość flawonoidów, 8- zawartość nieflawonoidowych fenoli, 9- zawartość
związków proantycyjaninowych, 10- intensywność koloru próbek, 11- barwa próbek,
12- stosunek transmitancji mierzonych dla rozcieńczonych próbek win przy 280 i 315
nm i 13- zawartość proliny) [55]. Dane można pobrać z [56].
Dane 3 dane zawierają wyniki analiz 124 próbek opium, które zebrano w trzech
prowincjach Indii (Madhya Pradesh, Uttar Pradesh i Rajasthan) [57]. W każdej próbce,
za pomocą chromatografii cieczowej, oznaczono zawartości piętnastu aminokwasów
takich jak cysteina, asparteina, treonina, seryna, kwas glutaminowy, glutamina,
alanina, walina, izoleucyna, leucyna, tyrozyna, fenyloalanina, histydyna, lizyna i
arginina. Dane, jak i dokładny opis procedury analitycznej znajduje się w [57].
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
26
Dane 4 zawierają profile stężeniowe ośmiu kwasów tłuszczowych 572 próbek oliwy
z oliwek [58]. Oliwki zebrano w dziewięciu regionach uprawnych Włoch (Kalabrii,
południowej Apulii, lądowej części Sardynii, nadmorskiej części Sardynii, wschodniej
Ligurii, zachodniej Ligurii i Umbrii). Zawartości poszczególnych kwasów
tłuszczowych (1- kwas palmitynowy, 2- kwas 3- oleopalmitynowy, 4- kwas
stearynowy, 5- kwas oleinowy, 6- kwas linolenowy, 7- kwas linolowy, 8- kwas
arachidowy oraz 9- kwas gadoleinowy) oznaczono za pomocą chromatografii gazowej.
Dane można pobrać z [59].
4.2 Wizualizacja struktury danych i badanie zależności pomiędzy zmiennymi
Bardzo cenną zaletą PCA jest umożliwienie wizualizacji wielowymiarowych danych
oraz ich interpretacji. Macierz wyników, T, oraz macierz wag, P, dostarczają bowiem
odpowiednio informacji o podobieństwach obiektów i zmiennych.
Zobaczmy, jak PCA pomaga w uzyskaniu informacji na temat struktury danych
i wzajemnych podobieństw pomiędzy próbkami. W tym celu posłużymy się drugim
zestawem danych. Ze względu na różnice w jednostkach, w jakich zmierzono
parametry, jak i ich różną skalę, przed analizą PCA dane zostały autoskalowane [23].
W przypadku danych, których zmienne autoskalowano, czynniki główne o
wartościach własnych mniejszych od jedności nie wnoszą istotnej informacji do opisu
danych [52].
Na Rys. 13a przedstawiono kumulacyjny procent wariancji danych dla pierwszych
dziesięciu czynników głównych. Kompresja danych metodą PCA nie jest zbyt
skuteczna, ponieważ pierwsze dwa czynniki opisują około 55,4% całkowitej wariancji
danych. Mimo to, z całego arsenału liniowych technik projekcji metoda PCA
zapewnia najlepszą kompresję danych. Pierwsze czynniki główne, modelujące
możliwie największą wariancję danych, najlepiej przedstawiają ich strukturę.
Aby ujawnić strukturę badanych danych i prześledzić ewentualne podobieństwa
pomiędzy próbkami win posłużymy się macierzą wyników. Na Rys. 13b,
przedstawiono położenie próbek w nowym układzie współrzędnych, zdefiniowanym
przez odpowiednie czynniki główne. Już pierwsze dwa czynniki główne pozwalają
ukazać niehomogeniczną strukturę danych (zob. Rys. 13b). Najczęściej, projekcje
wyników są źródłem informacji o tendencji danych do grupowania i/lub o próbkach,
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
27
które znacząco różnią się od pozostałych (tak zwanych obiektów odległych). Na
płaszczyźnie PC 1 - PC 2 można wyróżnić trzy grupy próbek - Rys. 13b. Przy analizie
poszczególnych projekcji wyników jako miarę podobieństwa pomiędzy próbkami
wykorzystuje się odległość euklidesową. Tak więc, próbki są tym bardziej do siebie
podobne pod względem chemicznym im mniejsze są pomiędzy nimi odległości
euklidesowe. Na Rys. 13c różnymi symbolami oznaczono, jaki gatunek wina
reprezentuje każda próbka. Grupy próbek nie są w pełni od siebie odseparowane, ale
można zobaczyć, iż grupują się one ze względu na rodzaje win. Zatem, możemy
wnioskować, iż pomiędzy gatunkami win istnieją wyraźne różnice ze względu na
wartości mierzonych parametrów fizyko-chemicznych.
Pierwsza grupa próbek win charakteryzuje się ujemnymi wartościami wyników
wzdłuż pierwszej osi i dodatnimi wzdłuż drugiej. Dla drugiej grupy próbek wartości
wyników przyjmują wzdłuż pierwszej osi zarówno ujemne jak i dodatnie wartości, a
wzdłuż drugiej ujemne. Natomiast wartości wyników próbek trzeciej grupy są
dodatnie wzdłuż obu osi.
Aby zbadać, które z parametrów są do siebie podobne, a które różnicują próbki win
dokonuje się projekcji wag na płaszczyzny zdefiniowane parami czynników głównych.
Wzajemne podobieństwa określa się na podstawie kąta, jaki tworzą pomiędzy sobą
dwa wektory wag o początku w punkcie [0 0] i końcach zdefiniowanych przez
odpowiednie wartości wag zmiennych na rozważanych projekcjach. Jeżeli kąt
pomiędzy dwoma parametrami jest bliski 00 wówczas są one silnie dodatnio
skorelowane. Kiedy kąt pomiędzy dwoma parametrami jest bliski 1800 to parametry
są silnie skorelowane, ale przeciwnie. Dwa parametry są niezależne (ortogonalne),
jeśli kąt pomiędzy nimi jest bliski 900. Dla analizowanych danych projekcje wag na
pierwsze dwa czynniki główne przedstawia Rys. 13d. Wynika z niego, iż znaczny
wkład do tworzenia pierwszego czynnika mają parametry 7 i 8 (flawonoidy i
nieflawonoidy), gdyż ich absolutne wartości wag są największe.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
28
a)
c)
100
3
80
2
70
PC 2 - 19,21%
procent opisanej wariancji danych
90
60
50
40
30
1
0
-1
-2
20
-3
Barolo
Grignolino
Barbera
10
0
-4
1
2
3
4
5
6
7
8
9
-4
10
-3
-2
d)
b)
0
1
2
3
4
wagi na czynniku głównym 2
2
1
0
-1
-2
-3
10
1
0.5
3
PC 2 - 19,21%
-1
PC 1 - 36,20%
kolejne czynniki główne
0.4
13
3
5
0.3
2
0.2
6
0.1
0
7
8
9
4
-0.1
12
-0.2
-4
11
-0.3
-4
-3
-2
-1
0
1
2
3
4
-0.4
-0.3
PC 1 - 36,20%
-0.2
-0.1
0
0.1
0.2
0.3
wagi na czynniku głównym 1
Rys. 13 a) Kumulacyjny procent wariancji danych 2 opisanej przez pierwsze dziesięć czynników
głównych, b) projekcja próbek na przestrzeń zdefiniowaną przez pierwsze dwa czynniki główne (obok
czynników głównych podano procent opisanej wariancji danych przez każdy czynnik), c) ta sama
projekcja, na której trzema symbolami oznaczono przynależność każdej próbki do jednego gatunku
wina: Barolo ({), Grignolino (Â) i Barbera () i d) projekcja wag na przestrzeń pierwszych dwóch
czynników głównych. Każdy symbol ({) przedstawia wartości wag takich parametrów jak:
1- alkohol, 2- kwas jabłkowy, 3- popiół, 4- zasadowość popiołu, 5- magnez, 6- całkowita zawartość
fenoli, 7- flawonoidy, 8- nieflawonoidowe fenole, 9- związki proantycyjaninowe, 10- intensywność
koloru, 11- barwa, 12- stosunek transmitancji mierzonych dla rozcieńczonych próbek win przy 280
i 315 nm i 13- prolina.
Z kolei te parametry praktycznie nie mają żadnego wkładu w konstrukcję drugiego
czynnika, gdyż wartości wag na drugim czynniku są bliskie zeru. Dla drugiego
czynnika największe znaczenie ma parametr 10 (intensywność koloru próbek). Z
projekcji wag wnioskujemy, iż parametry 6 i 7 są skorelowane dodatnio. Z tymi
parametrami są przeciwnie (ujemnie) skorelowane parametry 4 i 8. W praktyce
oznacza to, iż jeśli w badanych próbkach zawartość fenoli, flawonoidów i
proantycyjanianów rośnie, to zarazem obniża się zasadowość popiołów oraz
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
29
zawartość nieflawonoidowych fenoli. Aby zobrazować te zależności, na Rys. 14
przedstawiono relacje pomiędzy autoskalowanymi parametrami 6 i 7 oraz 7 i 8.
Dodatnia korelacja parametrów oznacza jednoczesny wzrost wartości obu parametrów,
a korelacja przeciwna, wzrost wartości jednego, a obniżenie wartości drugiego. Jeśli
kompresja danych metodą PCA nie jest efektywna, należy pamiętać o rozważnej
interpretacji zarówno projekcji wyników jak i wag, mając na uwadze, iż przedstawiają
one jedynie pewną część wariancji danych. Zatem ich analiza pozwala na
formułowanie bardzo ogólnych wniosków, a te powinny znaleźć odzwierciedlenie w
oryginalnych danych jak i dotychczasowej wiedzy o badanym problemie.
W przypadku danych 2, Rys. 14a świadczy o stosunkowo silnej dodatniej korelacji
pomiędzy parametrami 6 i 7 (współczynnik korelacji wynosi 0,86).
b)
3
nieflawonoidowe fenole (zmienna 8)
a)
flawonoidy (zmienna 7)
2.5
2
1.5
1
0.5
0
-0.5
-1
-1.5
2.5
2
1.5
1
0.5
0
-0.5
-1
-1.5
-2
-2
-1.5
-1
-0.5
0
0.5
1
1.5
całkowita zawartość fenoli (zmienna 6)
2
2.5
-1.5
-1
-0.5
0
0.5
1
1.5
2
2.5
3
flawonoidy (zmienna 7)
Rys. 14 Projekcje próbek na przestrzeń zdefiniowaną przez parametry: a) 6 (całkowita zawartość
fenoli) i 7 (flawonoidy) oraz b) 7 (flawonoidy) i 8 (nieflawonoidowe fenole).
Dla zmiennych 7 i 8, korelacja jest przeciwna, jak wskazuje projekcja wag, a jej
współczynnik wynosi zaledwie -0,54. Interpretując wagi parametrów rozważamy
jedynie ich projekcję wag na przestrzeń wybranych dwóch czynników głównych.
Najbardziej istotne z praktycznego punktu widzenia wydaje się być wskazanie
parametrów, które mają bezpośredni wpływ na obserwowaną strukturę widoczną na
projekcjach wyników. W tym celu należy równocześnie interpretować projekcje
wyników i wag, patrząc na ich wzajemne położenia na obu projekcjach. Dla
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
30
ułatwienia interpretacji, jeszcze raz, na Rys. 15 przedstawiono projekcje wyników i
wag pierwszych dwóch czynników głównych. Podczas interpretacji projekcji
wyników (opisujących określone próbki) jak i wag (opisujących zmienne) należy
uwzględnić ich znaki. W przypadku, gdy wyniki i wagi badanych próbek mają
ujemne lub dodatnie znaki ich iloczyn jest zawsze dodatni (zob. równanie 5). Dlatego
można powiedzieć, iż w takiej sytuacji dla próbek opisanych dodatnimi wartościami
wyników oraz dodatnimi wartościami wag lub ujemnymi wartościami wyników oraz
ujemnymi wartościami wag, określony parametr będzie miał relatywnie dużą wartość
w porównaniu z resztą obiektów. Skupmy się najpierw na grupie win Barbera. Z Rys.
15a wynika, iż te próbki opisane są dodatnimi wartościami wyników wzdłuż
pierwszej osi. Ponadto, projekcja wag (Rys. 15b) informuje o dużym wkładzie w
tworzenie pierwszego czynnika głównego parametrów 7 (flawonoidy) i 8
(nieflawonoidowe fenole). Są one opisane odpowiednio ujemną i dodatnią wartością
wagi. Możemy powiedzieć, iż w stosunku do innych próbek, w winach Barbera jest
relatywnie więcej fenoli, a także kwasu jabłkowego, a pH popiołów jest wyższe ze
względu na korelacje parametrów 2, 4 i 8. Ze względu na przeciwną korelację
parametru 7 z parametrem 8, wina Barbera mają małe zawartości flawonoidów. Wraz
z parametrem 7 podobną tendencję będą wykazywały parametry 6, 9 i 12, gdyż są one
ze sobą skorelowane.
W przypadku próbek win Barolo, tendencje obserwowane dla parametrów 7 i 8 są
przeciwne tych dla próbek win Barbera (zob. Rys. 15c i d). W próbkach tego wina
obserwuje się stosunkowo duże wartości parametrów 6, 7, 9 i 12 (ujemne wartości
wag i dodatnie odpowiednich wyników), zaś małe wartości parametrów 4 i 8
(dodatnie wagi i ujemne wartości wyników). Odmienność próbek win Grignolino
(ujemne wartości wyników wzdłuż drugiej osi) od pozostałych próbek win można
głównie tłumaczyć ich relatywnie mniejszą intensywnością koloru (parametr 10 –
dodatnia waga).
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
31
a)
c)
0.65
3
3
2
2
1
1
0.6
0.55
PC 2 - 19,21%
PC 2 - 19,21%
0.5
0
-1
0.45
0
0.4
-1
0.35
0.3
-2
-2
0.25
-3
-3
Barolo
Grignolino
Barbera
-4
-4
-3
0.2
0.15
-4
-2
-1
0
1
2
3
-4
4
-3
-2
13
3
5
0.3
2
0.2
6
0.1
0
7
8
9
4
-0.1
PC 2 - 19,21%
wagi na czynniku głównym 2
0.4
1
2
3
4
d)
10
1
0.5
0
PC 1 - 36,20%
PC 1 - 36,20%
b)
-1
5
3
4.5
2
4
3.5
1
3
0
2.5
-1
2
-2
1.5
12
-3
-0.2
11
-0.3
-0.4
-0.3
1
0.5
-4
-0.2
-0.1
0
0.1
wagi na czynniku głównym 1
0.2
0.3
-4
-3
-2
-1
0
1
2
3
4
PC 1 - 36,20%
Rys. 15 Projekcja próbek na przestrzeń zdefiniowaną przez pierwsze dwa czynniki główne, na której
trzema symbolami oznaczono typ wina: Barolo ({), Grignolino (Â) i Barbera (), b) projekcja wag na
przestrzeń pierwszych dwóch czynników głównych (każdy symbol przedstawia wartości wag takich
parametrów jak: 1- alkohol, 2- kwas jabłkowy, 3- popiół, 4- zasadowość popiołu, 5- magnez,
6- całkowita zawartość fenoli, 7- flawonoidy, 8- nieflawonoidowe fenole, 9- związki pro
antycyjaninowe, 10- intensywność koloru, 11- barwa, 12- stosunek transmitancji mierzonych dla
rozcieńczonych próbek win przy 280 i 315 nm i 13- prolina). Projekcja wyników pierwszych dwóch
czynników głównych, którym przypisano kolor proporcjonalny do wartości parametrów: c) 8 i d) 7.
W ten sposób nadaliśmy pełną interpretację projekcjom wyników tłumacząc, które z
oryginalnych zmiennych przyczyniają się najbardziej do obserwowanych grup win.
Interpretację wag autoskalowanych zmiennych ogranicza się zazwyczaj jedynie do
kilku wybranych zmiennych, których absolutne wartości wag są największe dla danej
projekcji.
Kolejnym ważnym aspektem analizy PCA jest identyfikacja grup parametrów, które
wnoszą do opisu danych podobną informację, co pozwala w uzasadnionych
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
32
przypadkach na ewentualną eliminację liczby mierzonych parametrów, obniżenie
kosztów analizy i skrócenie jej czasu.
4.3 Wpływ wstępnego przygotowania danych na konstrukcję czynników
głównych
Na przykładzie dwóch zestawów danych (dane 1 i 2) zademonstrujemy, że użyta
metoda wstępnego przygotowania danych do dalszej analizy ma duży wpływ na
konstrukcję czynników głównych i ich późniejszą interpretację. Na Rys. 16, dla
danych 1, przedstawiono projekcje wyników na płaszczyznę zdefiniowaną przez PC 1
i PC 2 odpowiednio dla oryginalnych widm, wycentrowanych oraz po transformacji
SNV i centrowaniu. Rys. 16a-c pokazują projekcje wyników oryginalnych danych 2
oraz danych tylko po centrowaniu i autoskalowaniu. Rezultatem zastosowania danej
metody wstępnego przygotowania danych jest zamiana odległości euklidesowych
pomiędzy obiektami w przestrzeni zmiennych, co przekłada się na wyjaśnioną przez
kolejne czynniki główne wariancję danych (zob. Rys. 16a-c) oraz na projekcje
obiektów. W przypadku danych 1, użycie transformacji SNV i centrowania pozwala
na ujawnienie w danych dwóch grup próbek, których obecność tłumaczy się różną
zawartością białka w zbożu. Dla danych 2, najlepsze wyniki uzyskano dla
autoskalowanych danych. Autoskalowanie umożliwiło wyeliminowanie dominacji
parametrów o względnie dużej wariancji. Na Rys. 16f widoczne są trzy grupy próbek,
które odpowiadają trzem gatunkom win.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
33
0.4
0.2
0.2
0
0
-0.2
-0.2
-0.4
-0.6
c)
0.6
0.4
-0.4
-0.6
-0.8
-0.8
1
0.5
PC 2 - 9,86%
b)
0.6
PC 2 - 3,69%
PC 2 - 0,03%
a)
0
-0.5
-1
-1
-1.2
-1.2
-1
-1.4
-1.4
-25
-24
-23
-22
-21
-20
-19
-18
-17
-5
-16
-4
-3
-2
20
60
10
40
0
20
0
-40
-40
-60
-50
-1200
-1000
-800
3
4
-3
-2
-1
-600
-400
-200
-60
-1000
PC 1 - 99,79%
0
1
2
3
PC 1 - 82,41%
f)
4
3
2
-20
-30
-1400
2
-10
-20
-1600
1
30
80
-80
-1800
0
PC 2 - 19,21%
e)
100
PC 2 - 0,17%
PC 2 - 0,21%
d)
-1
PC 1 - 95,92%
PC 1 - 99,96%
1
0
-1
-2
-3
-500
0
500
-4
-5
-4
-3
PC 1 - 99,81%
-2
-1
0
1
2
3
4
PC 1 - 36,20%
Rys. 16 Projekcje wyników na płaszczyznę zdefiniowaną przez pierwsze dwa czynniki główne dla:
a) oryginalnych danych 1, b) danych 1 po centrowaniu i c) danych 1 po transformacji SNV
i centrowaniu. Projekcje wyników na płaszczyznę zdefiniowaną przez pierwsze dwa czynniki główne
dla: d) oryginalnych danych 2, e) danych 2 po centrowaniu i f) danych 2 po autoskalowaniu.
4.4 Kompresja danych i częściowa redukcja szumu metodą PCA
PCA jest techniką kompresji danych i pozwala na częściową eliminację szumu. Aby
zilustrować te własności użyjemy pierwszego zestawu danych. Dane poddano
transformacji SNV [29] i centrowaniu by usunąć niepożądane efekty związane z
rozpraszaniem wiązki promieniowania elektromagnetycznego z zakresu bliskiej
podczerwieni na powierzchni próbek.
Efektywność kompresji danych metodą PCA można ocenić na kilka sposobów. Na
przykład, analizując wartości własne lub procent wariancji, jaki opisuje kilka
pierwszych czynników głównych (zob. Rys. 17a-c). Dla omawianych danych
interpretacja wartości własnych pierwszych ośmiu czynników głównych (Rys. 17a)
jak i kumulacyjnego procentu wariancji danych (Rys. 17c) pozwala wysnuć wniosek,
iż kompresja danych metodą PCA jest efektywna, gdyż pierwsze pięć czynników
głównych opisuje ponad 99,5% całkowitej wariancji danych. W celu ustalenia
optymalnej liczby czynników głównych, które zostaną użyte do późniejszej
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
34
5
rekonstrukcji danych, posłużono się kroswalidacją typu „wyrzuć jeden obiekt”. Rys.
16d przedstawia zależność RMSECV od liczby czynników głównych w modelu PCA.
Choć krzywa RMSECV nie osiąga wyraźnego minimum, to wykresy wartości
własnych i kumulacyjnego procentu opisanej wariancji danych pozwalają
wnioskować, iż cztery czynniki główne są znaczące. Każdy kolejny czynnik główny
niewiele wnosi do całkowitego opisu danych i dlatego następne czynniki główne
możemy utożsamić z szumem w danych lub błędem eksperymentalnym.
a)
c)
120
100
procent opisanej wariancji danych
90
wartość własna
100
80
60
40
20
80
70
60
50
40
30
20
10
0
1
2
3
4
5
6
7
0
8
1
2
indeks wartości własnej
b)
d)
90
5
6
7
8
2
1.6
70
1.4
60
RMSECV
procent opisanej wariancji danych
4
1.8
80
50
40
30
1.2
1
0.8
0.6
20
0.4
10
0
3
kolejne czynniki główne
-3
x 10
0.2
1
2
3
4
5
6
7
8
0
1
2
indeks czynnika głównego
3
4
5
6
7
8
kolejne czynniki główne
Rys. 17 a) Diagram pierwszych ośmiu wartości własnych, b) procent wariancji danych opisanej przez
każdy czynnik główny, c) kumulacyjny procent wariancji danych opisanej przez kolejne czynnik
główne oraz d) średni błąd kwadratowy kroswalidacji (RMSECV) jako funkcja liczby czynników
głównych w modelu PCA.
Teraz przedstawimy jak w oparciu o macierze wyników i wag można zrekonstruować
wyjściowe widma NIR. Ze względu na lepszą przejrzystość rysunków, na Rys. 18a-c
przedstawiono jedynie oryginalne widmo pierwszej próbki (oraz odpowiadające mu
zrekonstruowane widmo stosując modele PCA z 1, 2 i 4 czynnikami głównymi). Na
Rys. 18d-e pokazano reszty od tych modeli PCA dla wszystkich widm ze zbioru
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
35
danych. Im więcej czynników głównych w modelu PCA tym lepsza rekonstrukcja
widma pierwszej próbki.
1
0.5
0
-0.5
-1
-1.5
-2
c)
2
1.5
SNV-transformowana absorbancja
b)
2
1.5
SNV-transformowana absorbancja
SNV-transformowana absorbancja
a)
1
0.5
0
-0.5
-1
-1.5
1400
1600
1800
2000
2200
2400
1200
1400
1600
długość fali [mn]
0
-0.5
-1
-1.5
1800
2000
2200
1200
2400
0.1
0.05
0
-0.05
-0.1
-0.1
1400
1600
1800
2000
2200
2400
wartości reszt
0.1
0.05
-0.05
1800
2000
2200
2400
0.15
0.1
0
1600
długość fali [mn]
f)
0.15
0.05
1200
1400
długość fali [mn]
e)
0.15
wartości reszt
wartości reszt
d)
1
0.5
-2
-2
1200
2
1.5
0
-0.05
-0.1
1200
1400
1600
długość fali [mn]
1800
2000
długość fali [mn]
2200
2400
1200
1400
1600
1800
2000
2200
długość fali [mn]
Rys. 18 a-c) Widmo pierwszej próbki po SNV, oznaczone przerywaną linią oraz widmo
zrekonstruowane (ciągła linia) stosując model PCA z odpowiednio 1, 2 i 4 czynnikami głównymi; d-f)
reszty dla wszystkich próbek od modeli PCA odpowiednio z 1, 2 i 4 czynnikami głównymi.
W przypadku modelu PCA z czterema czynnikami różnice pomiędzy oryginalnym
widmem (przerywana linia), a widmem zrekonstruowanym (ciągła linia) są
praktycznie niezauważalne (zob. Rys. 18c).
Prześledźmy teraz zmiany w resztach od modelu PCA dla wszystkich widm próbek
zboża. Zauważamy tę samą tendencję, a mianowicie, ze wzrostem liczby czynników
użytych do rekonstrukcji widm, reszty od modelu PCA sukcesywnie maleją, a zatem
dane są coraz lepiej rekonstruowane (zob. Rys. 18d-f). Pomiędzy widmami NIR
zrekonstruowanymi stosując pierwsze cztery czynniki główne (optymalna liczba
czynników wyznaczona zgodnie z procedurą kroswalidacji), a oryginalnymi widmami
nie ma wizualnej różnicy, co pokazują Rys. 19a i b.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
36
2400
a)
b)
2
2
1.5
SNV-transformowana absorbancja
SNV-transformowana absorbancja
1.5
1
0.5
0
-0.5
-1
-1.5
1
0.5
0
-0.5
-1
-1.5
-2
-2
1200
1400
1600
1800
2000
długość fali [mn]
2200
2400
1200
1400
1600
1800
2000
2200
2400
długość fali [mn]
Rys. 19 a) Widma NIR próbek zboża po transformacji SNV oraz b) widma zrekonstruowane
używając cztery czynniki główne.
4.5 PCA, a obiekty odległe
Ponieważ czynniki główne są konstruowane tak, aby maksymalizować wariancję
projekcji, obecność w danych obiektów odległych wywiera silny wpływ na ich
konstrukcję. W przypadku obecności w danych obiektów odległych czynniki główne
modelują bardziej obiekty odległe, niż większość danych. Poszczególne projekcje
powinny ukazywać obiekty odległe, a zatem analiza czynników głównych powinna
umożliwiać ich detekcję. W wielu przypadkach na pierwszej projekcji (PC 1 – PC 2)
można zaobserwować obiekty o zdecydowanie odmiennych wartościach parametrów
w porównaniu z innymi obiektami. Należy jednak pamiętać, iż niektóre z obiektów
odległych nie zawsze będą widoczne na projekcjach wyników, ponieważ mogą mieć
jedynie duże wartości reszt od modelu PCA. Jeśli w danych są obiekty odległe,
odpowiednią techniką do eksploracji tych danych i identyfikacji obiektów odległych
jest stabilna metoda PCA. Konstrukcja stabilnych czynników głównych w tej
metodzie nie jest zaburzona obecnością obiektów odległych. Ponadto, stabilne
czynniki główne i reszty od stabilnego modelu mogą posłużyć do identyfikacji
obiektów odległych.
Na Rys. 20 pokazano jak silny wpływ wywierają obiekty odległe na tworzenie
poszczególnych kierunków w PCA na przykładzie symulowanych dwuwymiarowych
danych. Dane zawierają 100 obiektów wylosowanych z rozkładu normalnego.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
37
Współczynnik korelacji między zmiennymi wyniósł 0,8. Następnie, do danych
wprowadzono jeden obiekt odległy o współrzędnych [18 0]. Przed PCA dane
wycentrowano. Na Rys. 20a i b zaznaczono kierunki PC 1 i PC 2 maksymalizujące
wariancję danych, jeśli w danych odpowiednio nie ma i jest obiekt odległy.
a)
b)
3
PC 1
4
zmienna 2
1
zmienna 2
8
PC 1
6
2
0
PC 2
-1
PC 2
2
0
-2
-4
-2
-6
-3
-8
-4
-3
-2
-1
0
1
2
3
4
zmienna 1
0
5
10
15
zmienna 1
Rys. 20 Projekcje obiektów na płaszczyzny zdefiniowane przez pierwsze dwa czynniki główne
(PC 1 i PC 2), gdzie liniami oznaczono kierunki maksymalizujące wariancję symulowanych
dwuwymiarowych danych: a) bez obiektu odległego oraz b) z jedynym obiektem odległym.
Jak widać na Rys. 20, obecność jednego obiektu odległego może bardzo silnie
wpłynąć na tworzenie pierwszego czynnika głównego, a co za tym idzie i kolejnych.
Efekt ten jest tym istotniejszy im bardziej obiekt odległy różni się od pozostałych.
Omówimy teraz zastosowanie stabilnej metody PCA do diagnostyki obiektów
odległych na przykładzie danych 3. Mając na uwadze, że dane zawierają obiekty
odległe, a także, że parametry są w różnych zakresach, dane poddano stabilnemu
autoskalowaniu, tj. zamiast klasycznej średniej i odchylenia standardowego użyto ich
stabilne warianty – medianę i estymator skali Qn.
W stabilnej metodzie PCA, diagnostyka obiektów odległych w całości bazuje na
odległościach Mahalanobisa oraz resztach od stabilnego modelu o określonej
kompleksowości. Jednym ze sposobów oszacowania kompleksowości stabilnego
modelu PCA jest analiza wykresu stabilnych wartości własnych kolejnych stabilnych
czynników głównych. Wykres pierwszych dziesięciu stabilnych wartości własnych
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
38
wskazuje, że stabilny model PCA powinien zawierać sześć czynników (zob. Rys. 21a).
Na Rys. 21b przedstawiono projekcję próbek na płaszczyznę dwóch pierwszych
stabilnych czynników głównych. Widzimy, że z pewnością próbki 49 i 64 są
obiektami odległymi (zob. Rys. 21b). Na stwierdzenie, które z tych obiektów są
dobrymi obiektami odległymi, obiektami o dużych wartościach reszt od stabilnego
modelu bądź złymi obiektami odległymi pozwala analiza Rys. 21c. Na przykład
obiekty 49 i 64 (zob. Rys. 21c) to z pewnością złe obiekty odległe. Są one
stosunkowo daleko od większości obiektów w przestrzeni modelu, tj. mają duże
wartości reszt od modelu i duże odległości Mahalanobisa, dlatego będą wywierały
największy wpływ na czynniki główne, jeśli te skonstruujemy za pomocą klasycznego
modelu PCA. Oprócz złych obiektów odległych, diagram odległości pozwala na
wyróżnienie w analizowanych danych obiektów o dużych wartościach reszt od
stabilnego modelu PCA. Takim obiektem jest np. obiekt 88. Obiekty o dużych
wartościach reszt od stabilnego modelu nie są widoczne na projekcjach stabilnych
czynników głównych, gdyż po ich zrzutowaniu na przestrzeń pary stabilnych
czynników głównych „wpadają” one w obszar większości obiektów. Dlatego łatwo
można je błędnie utożsamić z regularnymi obiektami. Przykładem może być próbka,
której profil mierzonych parametrów ma inny kształt niż profile parametrów
pozostałych próbek. Dla dobrego opisu takiej próbki wymagana jest inna ilości
czynników głównych w modelu, niż bez niej. Kolejnym typem obiektów są tak zwane
dobre obiekty odległe. Charakteryzują się one stosunkowo dużymi odległościami
Mahalanobisa, lecz małymi resztami od stabilnego modelu. Tego typu obiekty są
jedynie daleko od większości danych w przestrzeni modelu. To np. próbki, dla
których profile parametrów charakteryzują się stosunkowo dobrą korelacją z profilami
innych próbek.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
39
a)
c)
z-transformowane reszty od modelu
stabilne wartości własne
12
10
8
6
4
2
0
30
49
25
15
36
10
5
0
1
2
3
4
5
6
7
8
9
0
10
10
15
20
25
30
5
88
0
stabilny PC 2
5
z-transformowane odległości Mahalanobisa
kolejne stabilne czynniki główne
b)
64
88
20
-5
36
-10
61
64
-15
49
-20
-45
-40
-35
-30
-25
-20
-15
-10
-5
0
5
stabilny PC 1
Rys. 21 a) Diagram przedstawiający kolejne stabilne wartości własne, b) projekcja obiektów na
przestrzeń dwóch pierwszych stabilnych czynników głównych oraz c) diagram obrazujący ztransformowane reszty od stabilnego modelu PCA względem z-transformowanych odległości
Mahalanobisa.
4.6 Konstrukcja czynników głównych dla danych z brakującymi elementami
Z różnych powodów analizowane dane mogą zawierać brakujące elementy. Wówczas
czynniki główne można konstruować stosując metodę EM-PCA. Dzięki niej buduje
się model PCA, podstawiając brakujące elementy tak, aby nie wywierały one wpływu
na model. Należy podkreślić, iż podstawianie brakujących elementów, np.
wartościami średnimi, jak to często ma miejsce, zaburza strukturę korelacyjną danych
i nie powinno być stosowane [51].
Dla zilustrowania działania metody EM-PCA posłużono się czwartym zestawem
danych, z którego losowo usunięto 3% całkowitej liczby elementów (tj. 137
elementów). Wzór brakujących elementów w macierzy danych przedstawiono na Rys.
22a. W celu wybrania optymalnej kompleksowości modelu analizowano wartości
własne. Optymalna liczba czynników w modelu EM-PCA, jaka powinna być użyta w
celu estymacji brakujących elementów, wynosi dwa (zob. Rys. 22b). Oprócz takiego
podejścia istnieją również inne, np. szybka kroswalidacja, której idee przedstawiono
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
40
w [60]. W przypadku EM-PCA, procedurę kroswalidacji (np. kroswalidacja typu
„wyrzuć jeden obiekt”) można stosować, aczkolwiek czas obliczeń jest znacząco
dłuższy. Na Rys. 22c przedstawiono dwie nałożone na siebie projekcje wyników na
płaszczyzny zdefiniowane przez dwa pierwsze czynniki główne, jakie otrzymano
stosując PCA dla kompletnych danych ({) oraz EM-PCA dla niekompletnych danych
(+), co pozwala na porównanie uzyskanych wyników. Pomimo różnic widocznych na
Rys. 22c, struktura danych z brakującymi elementami stosunkowo dobrze pokrywa
się z reprezentowaną przez pierwsze dwa czynniki główne dla kompletnych danych.
a)
c)
50
4
3
100
2
1
200
250
PC 2
ideks obiektu
150
300
0
-1
350
-2
400
450
-3
500
-4
550
1
2
3
4
5
6
7
8
-5
-6
ideks zmiennej
b)
-4
-2
0
2
4
6
PC 1
45
40
wartość własna
35
30
25
20
15
10
5
0
1
2
3
4
5
6
7
kolejny czynnik główny
Rys. 22 a) Schematyczna prezentacja macierzy danych (brakujące elementy zaznaczono na czarno),
b) diagram wartości własnych oraz c) projekcja obiektów na przestrzeń dwóch pierwszych czynników
głównych skonstruowanych dla (o) kompletnych danych metodą PCA oraz niekompletnych danych (+)
metodą EM-PCA.
Na jakość wyników uzyskanych z EM-PCA ma wpływ kilka czynników. Generalnie
można powiedzieć, iż zależy od rozkładu brakujących elementów w danych, ich ilości
oraz odpowiednio dobrej struktury korelacyjnej danych, to znaczy stosunkowo silnej
korelacji pomiędzy poszczególnymi zmiennymi. Odrębnym problemem jest analiza
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
41
danych, w których obecne są i brakujące elementy i obiekty odległe. To zagadnienie
szeroko omówiono w [61,62].
5. PODSUMOWANIE
Ze względu na swoje własności, analiza czynników głównych od ponad stu lat cieszy
się niegasnącą popularnością. Obecnie, PCA jest podstawowym narzędziem
eksploracji i kompresji macierzy danych (np. o wymiarach próbki × parametry).
Liczba publikacji opisujących zastosowania PCA jest ogromna. Choć PCA powstała z
myślą o analizie macierzy danych to coraz częściej jesteśmy zmuszeni prowadzić
eksplorację wielomodalnych danych. Przykładem takich danych są np. trójmodalane
dane, które powstają w trakcie monitorowania środowiska. Ich najczęstsze kierunki to
stacje pomiarowe × parametry × czas. W zależności od badanego problemu dane
mogą być N-modalne, a do ich eksploracji można użyć N-modalną analizę czynników
głównych [63,64,65,66].
6. LITERATURA
[1] J.N. Miller, J.C. Miller, Statistics and chemometrics for analytical chemistry,
Prentice Hall, London, 1999.
[2] D.L. Massart, B.G.M. Vandeginste, L.M.C. Buydens, S. de Jong, P.J. Lewi, J.
Smeyers-Verbeke, Handbook of chemometrics and Qualimetrics: part A, Elsevier,
Amsterdam, 1997.
[3] M. Daszykowski, B. Walczak, D.L. Massart, Projection methods in chemistry,
Chemometrics and Intelligent Laboratory Systems, 65 (2003) 97-112.
[4] S. Wold, K. Esbensen, P. Geladi, Principal component analysis, Chemometrics
and Intelligent Laboratory Systems, 2 (1987) 37-52.
[5] D.L. Massart, L. Kaufman, The interpretation of analytical chemical data by the
use of cluster analysis, R.E. Krieger Publishing Company, Florida, 1989.
[6] N. Bratchell, Cluster analysis, Chemometrics and Intelligent Laboratory Systems,
6 (1987) 105-125.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
42
[7] J.H. Friedman, J.W. Tukey, A projection pursuit algorithm for exploratory data
analysis, IEEE Transactions On Computers, 23 (1974) 881-890.
[8] A. Hyvärinen, J. Karhunen, E. Oja, Independent component analysis, John Willey
& Sons, New York, 2001.
[ 9 ] O.M. Kvalheim, N. Telnæs, Visualizing information in multivariate data:
applications to petroleum geochemistry. Part 1. Projection methods, Analytica
Chimica Acta, 191 (1986) 87-96.
[10] M. Daszykowski, From Projection Pursuit to other unsupervised chemometric
techniques, Journal of Chemometrics, 21 (2007) 270-279.
[11] G.P. Nason, Design and choice of projection indices, Ph.D. thesis, University of
Bath, 1992.
[ 12 ] D. Pena, F. Prieto, Cluster identification using projections, Journal of the
American Statistical Association, 96 (2001) 1433-1445.
[ 13 ] M. Daszykowski, I. Stanimirova, B. Walczak, D. Coomans, Explaining a
presence of groups in analytical data in terms of original variables, Chemometrics and
Intelligent Laboratory Systems, 78 (2005) 19-29.
[14] C. Croux, A. Ruiz-Gazen, A fast algorithm for robust principal components
based on projection pursuit, COMPSTAT: proceedings in Computational Statistics
(1996), 211-217, Heidelberg: Physica-Verlag.
[15] P. Gemperline, Practical guide to chemometrics, Taylor & Francis, London, 2006.
[16] D.L. Massart, Y. Vander Heyden, From tables to visuals: principal component
analysis, part 1, LC-GC Europe, 17 (2004) 586-591.
[17] D.L. Massart, Y. Vander Heyden, From tables to visuals: principal component
analysis, part 2, LC-GC Europe, 18 (2004) 84-89.
[18] K. Pearson, On lines and planes of closest fit to systems of points in space,
Philosophical Magazine, 6 (1901) 559-572.
[19] R. Fisher, W. MacKenzie, Studies in crop variation. II. The manurial response of
different potato varieties, Journal of Agricultural Science, 13 (1923) 311-320.
[20] H. Wold, Nonlinear estimation by iterative least squares procedures, in F. David
(Ed.), Research Papers in Statistics, Wiley, New York, 1966, pp. 411-444.
[21] H. Hotteling, Analysis of complex statistical variables into principal components,
Journal of Educational Psychology, 24 (1933) 417-441 and 498-520.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
43
[22] G.H. Golub, C.F. Van Loan, Matrix computations, The Johns Hopkins University
Press, Baltimore, 1996.
[23] B.G.M. Vandeginste, D.L. Massart, L.M.C. Buydens, S. de Jong, P.J. Lewi, J.
Smeyers-Verbeke, Handbook of chemometrics and qualimetrics: part B, Elsevier,
Amsterdam, 1998.
[24] W. Wu, D.L. Massart, S. de Jong, The kernel PCA algorithms for wide data. Part
I: Theory and algorithms, Chemometrics and Intelligent Laboratory Systems, 36
(1997) 165-172.
[ 25 ] H. Arodź, K. Rościszewski, Algebra i geometria analityczna w zadaniach,
Wydawnictwo Znak, Kraków, 2005.
[26] Q. Guo, W. Wu, D.L. Massart, C. Boucon, S. de Jong, Feature selection in
principal component analysis of analytical data, Chemometrics and Intelligent
Laboratory Systems, 61 (2002) 123-132.
[27] W.J. Krzanowski, Selection of variables to preserve multivariate data structure,
using principal components, Applied Statistics, 36 (1987) 22–33.
[ 28 ] I. Stanimirova, B. Walczak, D.L. Massart, Multiple factor analysis in
environmental chemistry, Analytica Chimica Acta, 545 (2005) 1-12.
[29] R.J. Barnes, M.S. Dhanoa, S.J. Lister, Standard normal variate transformation
and de-trending of near-infrared diffuse reflectance spectra, Applied Spectroscopy, 43
(1989) 772-777.
[30] E.R. Malinowski, Factor analysis in chemistry, John Wiley & Sons, New York,
1991.
[31] E.R. Malinowski, Theory of the distribution of error eigenvalues resulting from
principal component analysis with applications to spectroscopic data, Journal of
Chemometrics, 1 (1987) 33–40.
[32] E.R. Malinowski, Statistical F-tests for abstract factor analysis and target testing,
Journal of Chemometrics, 3 (1988) 49–60.
[33] R. Bro, K. Kjeldahl, A.K. Smilde, H.A.L. Kiers, Cross-validation of component
models: A critical look at current methods, Analytical and Bioanalytical Chemistry,
390 (2008) 1241-1251.
[ 34 ] W. Duch, J. Korbicz, L. Rutkowski, R. Tadeusiewicz, Sieci neuronowe,
Akademicka Oficyna Wydawnicza Exit, Warszawa, 2000.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
44
[35] M. Daszykowski, B. Walczak, D. L. Massart, Looking for natural patterns in
data: Part 1. Density-based approach, Chemometrics and Intelligent Laboratory
Systems, 56 (2001) 83-92.
[36] T. Næs, T. Isaksson, T. Fearn, T. Davies, A user-friendly guide to multivariate
calibration and classification, NIR Publications, Chichester, 2002.
[37] H. Martens, T. Næs, Multivariate calibration, Jon Wiley & Sons, Chichester,
1991.
[38] R. De Maesschalck, D. Jouan-Rimbaud, D.L. Massart, The Mahalanobis distance,
Chemometrics and Intelligent Laboratory Systems, 50 (2000) 1-18.
[ 39 ] A. de Juan, R. Tauler, Chemometrics applied to unravel multicomponent
processes and mixtures. Revisiting latest trends in multivariate resolution, Analytica
Chimica Acta, 500 (2003) 195-210.
[40] P.J. Rousseeuw, M. Debruyne, S. Engelen, M. Hubert, Robustness and outlier
detection in chemometrics, Critical Reviews in Analytical Chemistry, 36 (2006) 221242.
[41] S. Frosch Møller, J. von Frese, R. Bro, Robust methods for multivariate data
analysis, Journal of Chemometrics, 19 (2005) 549-563.
[42] N. Locantore, J.S. Marron, D.G. Simpson, N. Tripoli, J.T. Zhang, K.L. Cohen,
Robust principal component analysis for functional data (with comments), Test, 8
(1999) 1–74.
[43] K. Vanden Branden, M. Hubert, Robust classification in high dimensions based
on the SIMCA method, Chemometrics and Intelligent Laboratory Systems, 79 (2005)
10–21.
[44] I. Stanimirova, B. Walczak, D.L. Massart, V. Simeonov, A comparison between
two robust PCA algorithms, Chemometrics and Intelligent Laboratory Systems, 71
(2004) 83-95.
[45] R. Maronna, Principal components and orthogonal regression based on robust
scales, Technometrics, 47 (2005) 264-273.
[46] P.J. Rousseeuw, C. Croux, Alternatives to median absolute deviation, Journal of
the American Statistical Association, 88 (1993) 1273–1283.
[47] P.J. Huber, Robust statistics, John Wiley & Sons, Chichester, 1981.
[48] P.J. Rousseeuw, A.M. Leroy, Robust regression and outlier detection, John
Wiley & Sons, New York, 1987.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
45
[ 49 ] M. Daszykowski, K. Kaczmarek, Y. Vander Heyden, B. Walczak, Robust
statistics in data analysis - a review. Basic concepts, Chemometrics and Intelligent
Laboratory Systems, 85 (2007) 203-219.
[50] O. Hössjer, C. Croux, Generalizing univariate signed rank statistics for testing
and estimating a multivariate location parameter, Non-parametric Statistics, 4 (1995)
293-308.
[51] B. Walczak, D.L. Massart, Dealing with missing data. Part 1, Chemometrics and
Intelligent Laboratory Systems, 58 (2001) 15-17.
[ 52 ] K.G. Jöreskog, J.E. Klovan, R.A. Reyment, Methods in geomathematics,
Elsevier, Amsterdam, 1976.
[ 53 ] J.H. Kalivas, Two data sets of near infrared spectra, Chemometrics and
Intelligent Laboratory Systems, 37 (1997) 255-259.
[54] ftp://ftp.clarkson.edu/pub/hopkepk/Chemdata/Kalivas
[55] M. Forina, C. Armanino, M. Castino, M. Ubigli, Multivariate data analysis as a
discriminating method of the origin of wines, Vitis, 25 (1986) 189-201.
[56] http://michem.disat.unimib.it/chm/download/webdatasets/Wines.txt
[57] M.M. Krishna Reddy, P. Ghosh, S.N. Rasool, R.K. Sarin, R.B. Sashidhar, Source
identification of Indian opium based on chromatographic fingerprinting of amino
acids, Journal of Chromatography A, 1088 (2005) 158–168.
[ 58 ] M. Forina, C. Armanino, Eigenvector projection and simplified non-linear
mapping of fatty acid content of Italian olive oils, Annali di Chimica, 72 (1987) 127141.
[59] ftp://ftp.clarkson.edu/pub/hopkepk/Chemdata/Original/oliveoil.dat
[60] I. Stanimirova, B. Walczak, Classification of data with missing elements and
outliers, Talanta, 76 (2008) 602-609.
[61] I. Stanimirova, M. Daszykowski, B. Walczak, Dealing with missing values and
outliers in principal component analysis, Talanta, 72 (2007) 172-178.
[62] S. Serneels, T. Verdonck, Principal component analysis for data containing
outliers and missing elements, Computational Statistics and Data Analysis, 52 (2008)
1712-1727.
[ 63 ] R. Henrion, N-way principal component analysis theory, algorithms and
applications, Chemometrics and Intelligent Laboratory Systems, 25 (1994) 1-23.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
46
[ 64 ] P. Geladi, Analysis of multi-way (multi-mode) data, Chemometrics and
Intelligent Laboratory Systems, 7 (1989) 11-30.
[ 65 ] A. Smilde, R. Bro, P. Geladi, Multi-way analysis with applications in the
chemical sciences, John Wiley & Sons, Chichester, 2004.
[ 66 ] P.M. Kroonenberg, Applied multiway data analysis, John Wiley & Sons,
Hoboken, 2008.
M. Daszykowski, B. Walczak, Analiza czynników głównych i inne metody eksploracji danych,
w D. Zuba, A. Parczewski, Chemometria w analityce, IES, Kraków, 2008
47