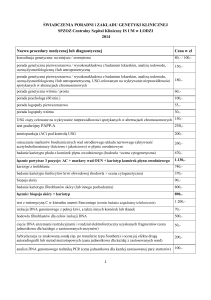
Bezpłatne badania genetyczne Wspólnie z firmą Genomed uruchomiliśmy program bezpłatnych badań genetycznych w zakresie czterech chorób rzadkich: fenyloketonurii, mukowiscydozy, choroby Gauchera oraz ceroidolipofuscynozy. Wszystkie osoby, które chciałyby skorzystać z badań prosimy o kontakt na adres [email protected] . Liczba badań jest ograniczona. Poniżej umieszczamy informacje na temat poszczególnych chorób oraz wskazań do wykonania badania. Fenyloketonuria jest najczęstszym wrodzonym, uwarunkowanym genetycznie zaburzeniem metabolizmu aminokwasów. Przyczyną choroby jest całkowity lub częściowy brak aktywności enzymu wątrobowego ‐ hydroksylazy fenyloalaninowej, katalizującego konwersję fenyloalaniny do tyrozyny. Wskazania do wykonania badania genetycznego: • diagnostyka hiperfenyloalaninemii wykrytej podczas badań przesiewowych noworodków; • diagnostyka przypadków upośledzenia umysłowego u dzieci z niedoborem masy ciała, małogłowiem, nieprawidłowym napięciem mięśniowym i zaburzeniami behawioralnymi, zwłaszcza występującymi rodzinnie lub z wyraźnym niedoborem melaniny w powłokach ciała; • diagnostyka przypadków niepowodzeń rozrodu, szczególnie jeśli kobieta nie może udowodnić ujemnego wyniku własnych badań przesiewowych, w tym nawracających poronień samoistnych (po wykluczeniu aberracji chromosomowych i trombofilii) lub urodzenia dziecka z niską masą urodzeniową, objawami uszkodzenia mózgowia i wadą serca; • badania rodzinne: wykrycie mutacji genu PAH u krewnego w rodzinach z hiperfenyloalaninemią, istnieje możliwość zastosowania w badaniach prenatalnych Mukowiscydoza to uwarunkowana genetycznie choroba wieloukładowa, manifestująca się głównie w obrębie układu oddechowego, układu pokarmowego oraz układu moczowo płciowego u mężczyzn. Wskazania do wykonania badania genetycznego • diagnostyka prenatalnie rozpoznanych przypadków hiperechogennego jelita, zwłaszcza przy prawidłowym kariotypie płodu oraz przypadków niedrożności smółkowej; • diagnostyka przypadków wysokiego poziomu immunoreaktywnego trypsynogenu wykrytego podczas badań przesiewowych noworodków; • diagnostyka przypadków przewlekłej choroby dróg oddechowych, zwłaszcza z przewlekłym kaszlem i zmniejszeniem pojemności płuc lub towarzyszącymi przewlekłymi zaburzeniami trawienia lub zwiększonym wydaleniem chlorków w pocie; • diagnostyka przypadków przewlekłej choroby układu pokarmowego, zwłaszcza z zaburzeniami trawienia i uszkodzeniem wątroby lub towarzyszącą przewlekłą chorobą dróg oddechowych lub zwiększonym wydaleniem chlorków w pocie; • diagnostyka przypadków niepłodności małżeńskiej w trakcie ustalania diagnozy lub przed przystąpieniem do procedur rozrodu wspomaganego medycznie, zwłaszcza w przypadkach podejrzenia niedrożności (obstrukcji) nasieniowodów, konieczne jest badanie obojga małżonków; • badania rodzinne: wykrycie mutacji genu CFTR u krewnego w rodzinach z przewlekłą chorobą dróg oddechowych, przewodu pokarmowego lub niepłodnością, istnieje możliwość zastosowania w badaniach prenatalnych. Choroba Gauchera jest chorobą metaboliczną zaliczaną do grupy lizosomalnych chorób spichrzeniowych. Choroba Gauchera jest powodowana przez recesywną zmianę w genie. Jeżeli dorosły człowiek, który posiada nieprawidłowy gen, ma partnera, który również jest nosicielem takiego samego genu, wówczas w przypadku ich dzieci istnieje ryzyko 1 do 4, że dziecko otrzyma dwie wadliwe kopie genu i wówczas będzie chore na tę chorobę. Prawdopodobieństwo, że rodzeństwo chorego dziecka będzie nosicielem choroby, wynosi 2 do 3. Wszystkie rodziny posiadające dzieci chore na tę chorobę powinny szukać pomocy i rady u swoich lekarzy lub specjalistów genetyki przed zaplanowaniem kolejnej ciąży. Wskazania do wykonania badania genetycznego • diagnostyka przypadków hepatosplenomegalii w wieku dziecięcym, zwłaszcza z towarzyszącymi zaburzeniami hemetologicznymi lub zahamowaniem rozwoju fizycznego lub występujących rodzinnie; • stwierdzenie charakterystycznych komórek Gauchera w biopsji szpiku kostnego; • diagnostyka przypadków nieimmunologicznego obrzęku płodu lub akinezji płodu z wielowodziem pojawiających się w 2 połowie ciąży, zwłaszcza kończących się wewnątrzmacicznym obumarciem płodu; • diagnostyka zespołu dziecka kolodionowego lub uogólnionej rybiej łuski z towarzyszącą hepatosplenomegalią lub dysmorfią; • badania rodzinne: wykrycie mutacji genu GBA u krewnego w rodzinach z powiększeniem narządów wewnętrznych i zaburzeniami hematologicznymi lub neurologicznymi, istnieje możliwość zastosowania w badaniach prenatalnych. Ceroidolipofuscynoza jest chorobą metaboliczną, zaliczaną do chorób spichrzeniowych, która atakuje komórki nerwowe organizmu. Ceroidolipofuscynozy stanowią heterogenną grupę wrodzonych chorób zwyrodnieniowych układu nerwowego, których wspólną cechą jest występowanie charakterystycznych złogów wewnątrzkomórkowych. Chociaż gromadzą się one w komórkach większości tkanek organizmu, objawy choroby dotyczą prawie wyłącznie ośrodkowego układu nerwowego. Ceroidolipofuscynoza może być wynikiem braku aktywności jednego z kilku enzymów i stąd rozróżniamy różne podtypy tej choroby, w tym najważniejsze ‐ ceroidolipofuscynozę niemowlęcą (brak aktywności enzymu tioesterazy palmityno – białkowej); ceroidolipofuscynozę poniemowlęca (wczesnodziecięcą) ‐ brak aktywności enzymu trójpeptydopeptydazy (TPP1), postać młodzieńczą (chorobę Battena) – mutacje w genie CLN3. Postaci wczesnodziecięca i młodzieńcza, które występują najczęściej, są równocześnie jednymi z najważniejszych przyczyn postępującego zwyrodnienia układu nerwowego u dzieci. Znanych jest ponad 50 mutacji w genie TPP1, odpowiedzialnych za ceroidolipofuscynozę wczesnodziecięcą (typu 2, CLN2). Najczęstsze patogenne warianty to p.R208X (c.622C>T) oraz c.509‐
1G>C (IVS5‐1G>C) ‐ stanowią one około 65% wszystkich mutacji występujących u pacjentów z CLN2. Ceroidolipofuscynoza jest powodowana przez recesywną zmianę w genie. Jeżeli dorosły człowiek, który posiada wadliwy gen, ma partnera, który również jest nosicielem takiego samego genu wówczas w przypadku ich dzieci istnieje ryzyko 1 do 4, że dziecko otrzyma dwie nieprawidłowe kopie genu i wówczas będzie chore na tę chorobę. Prawdopodobieństwo, że rodzeństwo chorego dziecka może być nosicielem choroby, wynosi 2 do 3. Wszystkie rodziny, posiadające dzieci chore na tę chorobę, powinny szukać pomocy i rady u swoich lekarzy lub specjalistów genetyki przed zaplanowaniem kolejnej ciąży.