NGS – ciąg dalszy
Bioinformatyczna analiza danych
Wykład 9
Dr Wioleta Drobik-Czwarno
Aplikacje NGS
Frese et al., 2013. Next-Generation Sequencing: From Understanding Biology to Personalized Medicine
NGS w medycynie
•
•
•
•
•
•
•
•
•
Onkologia
Zaburzenia hematologiczne
Warianty strukturalne
Wady wrodzone
Neurologia i psychiatria
Nieinwazyjne testy prenatalne
Kardiologia
Farmakogenomika
Choroby trudne w diagnostyce lub jeszcze niedokładnie
poznane
Demkow i Plonski, 2016. Clinical Applications for NextGeneration Sequencing.
Zalety NGS
•
•
•
•
•
Sekwencjonowanie wysoko-przepustowe – wiele próbek w jednym runie
Jednocześnie sekwencjonujemy wiele genów w wielu próbkach
Mniejszy koszt na pojedynczy gen
Różne rodzaje danych
Technologia jest cały czas udoskonalana
▫ Mniejszy koszt
▫ Mniejsze prawdopodobieństwo błędów
Wyzwania NGS
• Wymagająca analiza bioinformatyczna
• Duże ilości danych
• Technologia jest cały czas udoskonalana, co wiąże się ze ciągłymi zmianami
NGS a spersonalizowana terapia
onkologiczna
• Zrozumienie genetycznego podłoża nowotworów
• Diagnostyka nowotworów
• Przewidywanie reakcji na terapie (RNA-seq lub wykrywanie
wariantów DNA)
• Sekwencjonowanie krążącego DNA nowotworowego (ctDNA)
– DNA pochodzące od obumarłej komórki nowotworowej
• Wprowadzenie na szeroką skalę?
▫ Spadek cen
▫ Dopracowanie metod analizy bioinformatycznej
NGS a spersonalizowana terapia
onkologiczna
Cumminig et al. 2016.
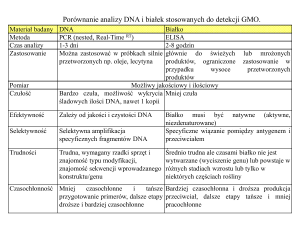
Zestawienie
za Shen i wsp., 2015
Największe problemy
• Dostępność próbek, a co za tym idzie często mała wielkość
próby
• Ograniczona ilość materiału a co za tym idzie DNA
▫ Materiał pochodzi z biopsji bądź jest to ctDNA
•
•
•
•
Konieczne porównanie ze zdrową tkanką
Problemy etyczne
Bezpieczeństwo danych
Brak dokładnej analizy ekonomicznej
Planowanie doświadczenia NGS
o czym należy pamiętać
• Losowy dobór próbek do eksperymentu
• Wykorzystanie istniejącej struktury rodzinowej
▫ Dziedziczenie
▫ Potwierdzenie wykrytych wariantów
• Możliwość sekwencjonowania w grupach (tzw. Pool-seq)
▫ Mniejsze koszty
▫ Prostsza analiza
Bioconductor
Czym jest Bioconductor?
• Projekt zapoczątkowany przez Roberta Gentlemana w 2001
roku
• Jest to repozytorium kodu służące do gromadzenia,
udostępniania i kontroli jakości narzędzi stworzonych w języku
R dotyczących bioinformatyki
Bioconductor
• Cele: zapewnienie
otwartej platformy do
analizy danych
genomowych, integracja
danych z różnych baz
danych, promocja dobrej
jakościowo
dokumentacji i
powtarzalności badań
• Istnieje możliwość
dodawania własnych
pakietów przez
użytkowników
Programowanie obiektowe
OOP ang. Object-oriented programming
• OOP: Przedstawienie świata rzeczywistego i relacji w nim
zachodzących za pomocą obiektów
• Zarówno R jak i Bioconductor opiera się paradygmacie
programowania OOP. Umożliwia to przedstawienie danych
biologicznych w formie obiektów o równych właściwościach.
• Najważniejsze pojęcia:
▫ Klasa – jest to typ zmiennej, inaczej definicja pewnej grupy obiektów.
Definiuje metody oraz atrybuty.
▫ Atrybut – jest to indywidualna cecha obiektów danej klasy
▫ Metoda – funkcjonalność dostarczana przez obiekty, inaczej funkcje,
które wykonują zadania na obiektach danej klasy
▫ Obiekt – jest instancją danej klasy, konkretna zmienna danego typu
Programowanie obiektowe
OOP ang. Object-oriented programming
• Przykład 1:
▫
▫
▫
▫
Klasa: Człowiek
Metody: spanie, jedzenie, poruszanie się, mówienie, itd.
Atrybuty: imię, nazwisko, data urodzenia itd.
Obiekty klasy człowiek: Janek, Kasia, Adam
• Przykład 2:
▫ Klasa: DNA
▫ Metody: zwróć sekwencję komplementarna, zwróć sekwencję
odwrotna, przetłumacz na sekwencję AA
▫ Atrybuty: długość, identyfikator
▫ Obiekty klasy DNA: sekwencja_1, sekwencja_2
Programowanie obiektowe
OOP ang. Object-oriented programming
• Funkcja generyczna – sprawdza podane argumenty i dobiera
odpowiednią metodę.
▫ Przykłady w R: plot, summary, print
▫ Wywołując funkję generyczną uzyskany efekt będzie zależny od
klasy jej argumentów – a dokładniej metod danej klasy
▫ Pozwala to dopasować obliczenia bezpośrednio do typu danych
Instalacja
• Zanim zaczniemy instalować podstawę oraz pakiety:
• source("http://bioconductor.org/biocLite.R")
• Instalacja
• biocLite() służy do instalacji szeregu pakietów tworzących
podstawę bioconductora
• Instalacja pakietow nie będących w podstawowej wersji
• biocLite("SNPchip")
Pakiety podstawowe do analizy
sekwencji
• Biostrings – klasy oraz funkcje dla sekwencji
• BSgenome i inne – sekwencje referencyjne dla różnych
gatunków
• GenomicRanges – manipulowanie przedziałami genomowymi
• GenomicFeatures – pozyskiwanie i manipulowanie danymi z
publicznych baz danych
Pakiety podstawowe
• Biobase:
▫ Podstawowe klasy i definicje, które pozwalają na dokładny
opis danych oraz manipulowanie nimi
▫ Funkcje niezbędne do działania wielu pakietów, zastępujące
funkcję w podstawowej wersji R
• Huber, W., Carey, J. V, Gentleman, R., Anders, S., Carlson, M., Carvalho, S. B, Bravo, C. H,
Davis, S., Gatto, L., Girke, T., Gottardo, R., Hahne, F., Hansen, D. K, Irizarry, A. R, Lawrence, M.,
Love, I. M, MacDonald, J., Obenchain, V., Ole's, K. A, Pag'es, H., Reyes, A., Shannon, P., Smyth,
K. G, Tenenbaum, D., Waldron, L., Morgan and M. (2015). “Orchestrating high-throughput
genomic analysis with Bioconductor.” Nature Methods, 12(2), pp. 115–121.
Pakiety podstawowe
• Biostrings:
▫ Wydajne (oszczędzające RAM) kontenery na ciągi znaków,
algorytmy dopasowania oraz inne funkcje, dla szybkiego
manipulowania dużymi sekwencjami biologicznymi lub ich
zestawami.
Pagès H, Aboyoun P, Gentleman R and DebRoy S (2017). Biostrings: String objects
representing biological sequences, and matching algorithms. R package version 2.44.0.
Przykładowe pakiety:
•
Adnotacja: annotate, annaffy, biomaRt
•
Graphics/GUIs: geneplotter, hexbin, limmaGUI, exploRase
•
Pre-processing: affy, affycomp, oligo, makecdfenv, vsn, gcrm, limma
•
Differential gene expression: genefilter, limma, ROC, siggenes, EBArrays,
factDesign
•
GSEA/Hypergeometric Testing GSEABase, Category, GOstats, topGO
•
Graphs and networks: graph, RBGL, Rgraphviz
•
Flow Cytometry: flowCore, flowViz, flowUtils
•
Protein Interactions: ppiData, ppiStats, ScISI, Rintact
•
Sequence Data: Biostrings,ShortRead,rtracklayer,IRanges,GenomicFeatures,
VariantAnnotation
•
Other data: xcms, DNAcopy, PROcess, aCGH, rsbml, SBMLR, Rdisop
Instrukcje
Źródło: https://www.bioconductor.org/help/workflows/
I dużo więcej w advanced workflows…
GWAS w R a Bioconductor
• GenABEL
• Bioconductor
▫ GWASTools
▫ SNPRelate
SNPRelate umożliwia wykonanie analizy PCA oraz analizę
podobieństwa
•PCA vs MDS:
• informacja o % zmienności genetycznej wyjaśnianej przez każdy
komponent
•Informacje o % udziale każdego markera w danych komponencie
(loadings)
Jak zacząć z pakietem Biostrings?
• Instalacja:
▫ source("http://bioconductor.org/biocLite.R")
▫ biocLite("Biostrings”)
• Ładowanie pakietu:
▫ require(Biostrings)
• Wygenerowanie przykładowej sekwencji o długości 10pz:
seq_raw <- sample(LETTERS[c(1,3,7,20)], size=10, replace=TRUE)
Wynik:
[1] "C" "G" "T" "A" "T" "T" "G" "G" "A" "A"
seq = paste(seq_raw, collapse="")
Klasa Xstrings i BStrings
• Klasa pozwala na tworzenie, przechowywanie oraz zapewnia
metody pracy z różnymi typami tekstu
• Najważniejsze subklasy – Bstring - DNAString, RNAString,
AAString
• DNAString – klasa obiektów, umożliwia wydajne
manipulowanie i przechowywanie długich sekwencji DNA
• Bstring przechowuje sekwencje w kodzie IUPAC
• Tworzenie obiektu klasy Bstring
bstring<-Bstring(„tekst”)
bstring
9-letter „Bstring” instance
seq: tekst
IUPAC
Międzynarodowa Unia Chemii
Czystej i Stosowanej
ang. International Union of
Pure and Applied Chemistry
• Kod, który uwzględnia
również zdegenerowane
zasady
Dziękuję za uwagę