Zakład Chemii Środowiska
Panel Chemia Środowiska
Pracownia Specjalizacyjna
Ćwiczenie 06
Oznaczanie całkowitej zawartości oraz
biodostępności wybranych metali cięŜkich metodą
płomieniowej atomowej spektrometrii
absorpcyjnej (F-AAS)
Prowadzący: Dr Paweł Miśkowiec ([email protected])
Miejsce wykonywania ćwiczenia:
Zakład Chemii Środowiska, Wydział Chemii UJ
ul. Gronostajowa 3 (III Kampus UJ), pok. 007, 015.
© Paweł Miśkowiec, ZCHŚ UJ
Wstęp
Atomowa spektrometria absorpcyjna (ASA) jest techniką analityczną, której zadaniem jest
oznaczenie zawartości wybranych pierwiastków chemicznych (głównie metali) w próbkach
ciekłych, gazowych oraz stałych. Zasada pomiaru opiera się na zjawisku absorpcji promieniowania
o specyficznej długości fali przez wolne atomy pierwiastka. Zastosowanie atomowej spektrometrii
absorpcyjnej jest bardzo szerokie, od ochrony środowiska, poprzez analizę składu, chemię sądową
aŜ do szeroko pojętych badań biologicznych, biochemicznych i medycznych.
Podstawy metody i aparatura
Spektroskopia jest nauką o powstawaniu i interpretacji widm powstających w wyniku
oddziaływania promieniowania na materię. W przypadku oddziaływania promieniowania na atomy
mamy do czynienia ze spektrometrią atomową.
Metody, których podstawą są widma atomowe moŜna najogólniej podzielić na metody absorpcyjne
i emisyjne. Przykładem pierwszej jest absorpcyjna spektrometria atomowa (ASA, ang. AAS).
Do drugich naleŜą np. historycznie pierwsza technika wzbudzania atomów: fotometria płomieniowa
oraz tak nowoczesna jak plazmowa emisyjna spektrometria atomowa (ICP-AES).
Metody spektrometrii atomowej oparte są na przejściach elektronów pomiędzy określonymi
poziomami energetycznymi. W standardowych warunkach temperatury i ciśnienia atomy znajdują
się w elektronowym stanie podstawowym, nie mają więc moŜliwości emitowania promieniowania.
Elektrony będące w stanie podstawowym mogą jednakŜe pochłonąć promieniowanie o określonej
długości fali i energii równej róŜnicy poziomów elektronowych w atomie. Następuje zjawisko
absorpcji będące podstawą metod absorpcyjnych. Atom znajduje się więc w stanie wzbudzonym.
Kolejnym etapem jest przejście elektronu na niŜszy poziom energetyczny, czemu towarzyszy
emisja promieniowania o częstotliwości: ν = (E1 – E0)/h, gdzie E1 energia atomu na poziomie
wzbudzonym, E0 energia atomu w stanie podstawowym, h – stała Plancka. Emisja promieniowania
przez atomy będące w elektronowym stanie wzbudzonym jest podstawą metod emisyjnych.
Podstawy teoretyczne powstawania widm atomowych najczęściej przedstawia się na przykładzie
najprostszego widma – atomu wodoru. Widmo to składa się z szeregu tzw. linii widmowych1
układających się w zespoły zwane seriami. W seriach linie widmowe zagęszczają się w miarę
przechodzenia w kierunku fal krótszych. Przejście elektronu w atomie wodoru związane z emisją
bądź absorpcją promieniowania wiąŜe się ze zmianą jego głównej liczby kwantowej n. Długość fali
(λ) (a co za tym idzie energię promieniowania), jaką naleŜy dobrać, aby nastąpiło przejście
elektronu, bądź teŜ długość fali promieniowania emitowanego na skutek przejścia odwrotnego
moŜna wyrazić wzorem:
1/λ = R (1/n12 – 1/n22),
gdzie n1 i n2 to główne liczby kwantowe poziomów energetycznych, pomiędzy którymi nastąpiło
przejście elektronu, a R to stała Rydberga wynosząca 0,0109677 nm-1.
Długości fal odpowiadające liniom widmowym tych samych serii moŜna znaleźć podstawiając
tę samą wartość n1 oraz róŜne wartości n2.
1
Linia widmowa, względnie wąski obszar w widmie emisyjnym lub absorpcyjnym atomu lub związku chemicznego,
odpowiadający częstości fali emitowanej lub absorbowanej przez dane indywiduum chemiczne.
2
© Paweł Miśkowiec, ZCHŚ UJ
Równania dla długości fal poszczególnych serii widmowych atomu wodoru wyglądają więc
następująco:
1. Seria Lymana
1/λ = R (1/12 - 1/n2), n = 2, 3, 4,…,
2. Seria Balmera
1/λ = R (1/22 - 1/n2), n = 3, 4, 5,…,
3. Seria Paschena
1/λ = R (1/32 - 1/n2), n = 4, 5, 6,…,
4. Seria Bracketa
1/λ = R (1/42 - 1/n2), n = 5, 6, 7,…,
… itd. (rys.1)
Energia
Przeprowadzając odpowiednie obliczenia dla najbardziej prawdopodobnych przejść z poziomów
n+1 oraz n+2 (czyli odpowiednio linii α oraz β) otrzymujemy:
seria Lymana: 122 nm i 103 nm,
seria Balmera: 656 nm i 486 nm – zakres widzialny,
seria Paschena: 1876 nm i 1282 nm,
seria Bracketa: 4052 nm i 2626 nm,
Rys. 1. Cztery pierwsze serie widmowe atomu wodoru.
3
© Paweł Miśkowiec, ZCHŚ UJ
Absorpcyjna spektrometria atomowa
Absorpcyjna spektrometria atomowa jest oparta na zjawisku absorpcji promieniowania
elektromagnetycznego przez swobodne atomy.
Podstawę metody moŜna w skrócie ująć w następujących punktach:
1. Źródłem analizowanych linii absorpcyjnych są atomy swobodne.
2. Swobodne atomy mogą absorbować promieniowanie o długościach fali, które mogą
emitować.
3. Otrzymane widmo absorpcyjne jest charakterystyczne dla danego rodzaju atomów.
W metodzie ASA badając absorpcję promieniowania przez swobodne atomy wykorzystuje się tzw.
plazmę niskotemperaturową (do 4000 K). Liczba swobodnych atomów w plazmie jest wprost
proporcjonalna do stęŜenia roztworu badanego. Absorbancja A natomiast w tej metodzie zaleŜna
jest liniowo od liczby swobodnych atomów. Biorąc powyŜsze fakty pod uwagę moŜna zapisać:
A=ε·b·N
gdzie: ε - molowy współczynnik absorpcji (wielkość charakterystyczna dla danego rodzaju atomów
i określonej długości fali), b - długość drogi optycznej, N - ilość wolnych atomów na drodze
promieniowania.
Ilość wolnych atomów N moŜna zamienić na proporcjonalnie z nią związane stęŜenie atomów (c)
w próbce, co w stałych warunkach pomiaru dla określonej długości fali daje liniową zaleŜność:
A=a·c
gdzie: a – współczynnik proporcjonalności.
Linie widmowe mają kształt krzywych Gaussa, a ich szerokość moŜna wyrazić wzorem:
∆λ = 1/τ2 - 1/τ1
gdzie τ2 jest czasem Ŝycia stanu wzbudzonego a τ1 czasem Ŝycia stanu podstawowego. Zawsze
jednak obserwujemy pewne poszerzenie linii widmowych, na które wpływają właściwości plazmy,
m.in. jej temperatura i ciśnienie. Wpływ temperatury tłumaczy się Efektem Dopplera polegającym
na tym, Ŝe obiekt absorbujący porusza się względem źródła emisji i detektora (gdy atom porusza się
w stronę detektora, ν wzrasta i λ maleje, gdy atom oddala się od detektora, ν maleje i λ wzrasta).
Wzrost natomiast ciśnienia gazu obojętnego (Efekt Lorenza) powoduje zwiększoną liczbę zderzeń
między atomami, co skraca czas Ŝycia stanu wzbudzonego i zgodnie z zasadą nieoznaczoności
Heisenberga, powoduje poszerzenie linii widmowych.
Wykorzystując liniową zaleŜność absorbancji od stęŜenia w metodzie ASA (jak i AES) pomiary
ilościowe prowadzi się zazwyczaj metodą serii wzorców, jak równieŜ metodą dodatku wzorca.
Aparatura
Zasadniczymi elementami spektrometru absorpcji atomowej są (rys.2.):
- źródło promieniowania,
- układ wprowadzania próbki,
- atomizer,
- monochromator,
- detektor,
- wzmacniacz,
- rejestrator (komputer).
4
© Paweł Miśkowiec, ZCHŚ UJ
Rys. 2. Schemat blokowy spektrometru absorpcji atomowej.
Jako źródła promieniowania stosuje się dwa rodzaje lamp:
1. lampy z katodą wnękową (HCL),
2. lampy z wyładowaniem bezelektrodowym (EDL).
Lampa HCL jest rurką wypełnioną neonem lub argonem, w której znajduje się anoda wykonana
z wolframu oraz katoda wnękowa wykonana z metalu, którego linie rezonansowe2 lampa ma
emitować. W lampie takiej wzbudzone dodatnio naładowane jony gazu szlachetnego bombardują
katodę i wybijają z niej atomy metalu w procesie określanym często rozpylaniem jonowym. Wybite
atomy w stanie gazowym ulegają wzbudzeniu, emitując następnie charakterystyczne
promieniowanie. W przypadku kilku pierwiastków, jak As, Sb, Se, Te, nie moŜna zbudować lampy
HCL, dlatego stosuje się lampy EDL, gdzie wzbudzenie atomów metalu następuje na skutek
działania pola elektromagnetycznego o duŜej częstości.
Zadaniem atomizerów jest wytworzenie swobodnych atomów danego pierwiastka z próbki
analitycznej. NajwaŜniejszymi rodzajami atomizerów są:
- atomizery płomieniowe,
- atomizery elektrotermiczne,
- atomizery wodorkowe,
- atomizery wykorzystujące zimne pary rtęci.
W atomizerach płomieniowych przejście od roztworu do gazu atomowego odbywa się poprzez :
- nebulizację, czyli rozpylenie analizowanego roztworu i doprowadzenie go w postaci mgły
do płomienia,
- atomizację w płomieniu palnika, gdzie gazem utleniającym jest powietrze, tlen lub N2O a gazem
palnym acetylen, gaz świetlny lub propan-butan. W najczęściej stosowanym układzie
powietrze-acetylen, temperatura płomienia wynosi ok. 2300 °C.
Szczegółowo procesy zachodzące w atomizerze przedstawia poniŜszy schemat:
M+
rozpylenie
desolwatacja
topnienie
parowanie
atomizacja (dysocjacja termiczna)
wzbudzenie
jonizacja
reakcje syntezy
2
+
A−
(roztwór)
M+
+
A−
(aerozol)
MA
(ciało stałe)
MA
(ciecz)
MA
(gaz)
M0
+
A0
(gaz)
M*
(gaz)
M+
+
e−
(gaz)
M + O → MO i inn.
Linia rezonansowa - linia widmowa związana z powrotem elektronu z poziomu rezonansowego
(najniŜszego poziomu wzbudzonego) na poziom podstawowy.
5
© Paweł Miśkowiec, ZCHŚ UJ
NajwaŜniejszą z punktu widzenia analizy reakcją w płomieniu jest dysocjacja termiczna,
dostarczająca swobodnych atomów. Inne reakcje, takie jak: jonizacja, wzbudzenie, czy reakcje
syntezy obniŜają liczbę wolnych atomów, a zatem i czułość metody.
W atomizerach elektrotermicznych stosuje się kuwety grafitowe. Podczas ogrzewania próbki
następuje jej osuszenie, spopielenie i w fazie trzeciej – atomizacja.
W technice atomizacji wodorkowej wytwarza się lotne wodorki metali, które ulegają rozkładowi
termicznemu do swobodnych atomów w kuwecie absorpcyjnej. Technikę tę stosuje się do
pierwiastków, które trudno przeprowadzić w stan pary, przede wszystkim grup 14-16 układu
okresowego pierwiastków, takich jak Sn, Se, As.
Szczególnym przypadkiem pod względem atomizacji jest rtęć. Jest ona jedynym metalem
charakteryzującym się wyraźną pręŜnością pary w temperaturze pokojowej, nie reagując
jednocześnie praktycznie z tlenem atmosferycznym. Dzięki temu, po redukcji do rtęci metalicznej,
moŜna oznaczać ją w postaci pary w kuwecie przepływowej. Technika ta zwana jest techniką
zimnych par.
Monochromatory
Monochromator umoŜliwia wydzielenie promieniowania o jednej długości fali zwanej linią
rezonansową i przepuszczenie jej do detektora. W metodzie ASA jest to zazwyczaj układ szczelin
i ruchomych zwierciadeł, dzięki którym moŜna wyizolować z widma szukaną długość fali.
Detektory
Zarówno w metodzie ASA jak i AES do pomiaru natęŜenia promieniowania słuŜą fotopowielacze,
wzmacniające i przekształcające sygnał w postać cyfrową, w której to trafia on do komputera.
UŜywany w ćwiczeniu absorpcyjny spektrometr atomowy AAnalyst 300 firmy Perkin Elmer
wyposaŜony jest w:
- automatyczny palnik,
- programowalny układ kontroli płomienia i przepływu gazu,
- automatyczny układ selekcji lamp przeznaczony do analiz wielopierwiastkowych,
- dwuwiązkowy system optyczny,
- automatyczny układ monochromatyczny kontroli długości fali i szerokości szczeliny,
- deuterowy korektor tła,
- sterowane przez centralny komputer systemy atomizacji i kontroli oprzyrządowania.
6
© Paweł Miśkowiec, ZCHŚ UJ
Mineralizacja
Mineralizacja polega na rozkładzie i utlenieniu związków organicznych zawartych w próbce
i przeprowadzeniu pozostałych składników do roztworu. Główne sposoby mineralizacji to
mineralizacja na sucho oraz mineralizacja na mokro.
Do grupy technik rozkładu próbek na sucho naleŜą między innymi:
spopielanie,
mineralizacja niskotemperaturowa w plazmie tlenowej,
mineralizacja w tlenie,
stapianie.
Mineralizację na mokro moŜna prowadzić w systemach otwartych lub zamkniętych
(ciśnieniowych):
z zastosowaniem konwencjonalnego ogrzewania,
z wykorzystaniem ultradźwięków,
promieniami UV,
z wykorzystaniem energii mikrofalowej.
Rozkład matrycy odbywa się za pomocą jednego lub kilku mocnych kwasów mineralnych, czasami
z dodatkiem innych związków o właściwościach utleniających. Utleniaczami są zazwyczaj HNO3,
H2SO4, HCIO4 oraz H2O2.
Obecnie jedną z popularniejszych metod rozkładu próbki jest zastosowanie mineralizacji
ciśnieniowej wspomaganej energią mikrofalową. Technika ta powstała przez połączenie dwóch
innych: mineralizacji ciśnieniowej w tzw. ,,bombach teflonowych” oraz mineralizacji przy uŜyciu
energii mikrofal. W przypadku omawianej mineralizacji ciśnieniowej procesy rozkładu są
dodatkowo wspomagane oddziaływaniem mikrofalowego promieniowania elektromagnetycznego
o częstotliwości 2450 MHz. Promieniowanie jest bezpośrednio absorbowane przez polarne
cząsteczki np. wodę czy kwasy nieorganiczne. Jest to duŜo efektywniejsze i szybsze źródło energii
niŜ ogrzewanie klasyczne oparte na konwekcji i przewodnictwie cieplnym naczynia. Analizowana
próbka reaguje z kwasami mineralnymi w podwyŜszonej temperaturze w zamkniętym naczyniu.
Powstające w wyniku wydzielania gazów ciśnienie, umoŜliwia stosowanie wyŜszych temperatur,
niŜ temperatury wrzenia kwasów w układach otwartych, a zatem skraca czas reakcji. Naczynia
stosowane w mineralizacji mikrofalowej muszą być wykonane z materiału przepuszczającego
promieniowanie mikrofalowe i jednocześnie odpornego na działanie stęŜonych kwasów. Obecnie
najczęściej uŜywa się teflonu oraz kwarcu.
Zaletami mineralizacji mikrofalowej są:
dobra powtarzalność,
stosunkowo prosta metodyka analizy,
krótki czas przebiegu procesu,
niewielkie zuŜycie odczynników,
moŜliwość oznaczania w mineralizacji wielu pierwiastków w szerokim zakresie stęŜeń,
ograniczone straty analitu,
ograniczona moŜliwość zanieczyszczenia próbki,
moŜliwość roztwarzania niewielkich odwaŜek analizowanego materiału (nawet rzędu 0,1 g),
stosunkowo proste dostosowanie parametrów decydujących o mineralizacji (rodzaju i stęŜenia
kwasów, mocy generatora, ciśnienia) do rodzaju analizowanych próbek.
Wady mineralizacji mikrofalowej to przede wszystkim:
wysoka cena aparatu,
maksymalna masa mineralizowanej próbki w większości przypadków nie moŜe przekraczać 5 g,
mała przepustowość mineralizacji, w przypadku aparatu jednostanowiskowego.
7
© Paweł Miśkowiec, ZCHŚ UJ
Metale cięŜkie w środowisku
Termin metale cięŜkie nie jest ściśle zdefiniowany. W naukach biologicznych pojęcie to uŜywane
jest często w odniesieniu do pierwiastków metalicznych oraz metaloidów odznaczających się
toksycznością dla środowiska, w tym szczególnie dla człowieka. Jedna z szerzej akceptowanych
definicji, określa metale cięŜkie jako pierwiastki metaliczne i metaloidy o liczbie atomowej wyŜszej
niŜ 20 i gęstości większej niŜ 4,5g/cm3. Zgodnie z tak przyjętą definicją moŜemy wyróŜnić ok. 65
metali cięŜkich. Najbardziej znanymi pierwiastkami tej grupy są: As, Bi, Cd, Co, Cr, Cu, Fe, Hg,
Mn, Mo, Ni, Pb, Sn, Zn. Spośród wymienionych jedne są mikroelementami – pierwiastkami
niezbędnymi do Ŝycia dla organizmów Ŝywych w niewielkich ilościach (np. Cr, Zn, Cu), inne zaś są
szkodliwe dla organizmów, niezaleŜnie od stęŜenia, gdyŜ zaburzają podstawowe procesy
metaboliczne (np. Cd, Hg, Pb). NajwaŜniejsze szkodliwe skutki oddziaływania na organizm
człowieka wybranych metali cięŜkich oraz ich najwyŜsze dopuszczalne stęŜenia w glebach róŜnie
uŜytkowanych przedstawia tabela 1.
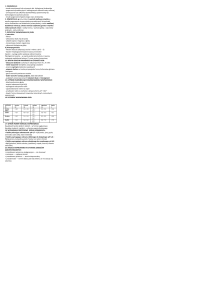
Tabela 1. Wpływ wybranych metali cięŜkich na organizm człowieka oraz dopuszczalne wartości
stęŜeń metali w glebie lub ziemi wg Rozporządzenia Ministra Środowiska z dn. 9.09.2002.
Metal
Cd
Pb
Hg
Zn
Cu
Ni
Cr
Skutki szkodliwego działania na organizm
Odwapnienie i deformacja kości, zanik mięśni (choroba
„itai-itai”) i węchu, nadciśnienie, nowotwory płuc,
gruczołów rodnych, jamy ustnej.
Bóle głowy, osłabienie pamięci, agresja, otępienie
umysłowe, zaburzenia psychiczne, uszkodzenie nerek.
Upośledzenie narządów zmysłów u dzieci niedorozwój
umysłowy (choroba z Minamaty), u płodu niedorozwój
mózgu, zmysłów, paraliŜ kończyn, drŜenie rąk i nóg, paraliŜ
mowy.
Niedobór – łysienie, karłowatość, ograniczenie funkcji
rozrodczych.
Nadmiar – niedokrwistość, obniŜenie przyswajalności
innych niezbędnych pierwiastków.
Niedobór – pogorszenie wchłaniania Ŝelaza przez
organizm i zmniejszenie liczby czerwonych krwinek.
Nadmiar – zaburzenia w pracy układu pokarmowego,
uszkodzenia wątroby czy nerek.
Niedobór – zmiana pigmentacji, zniekształcenia kości,
obrzęk stawów, zwyrodnienie wątroby.
Nadmiar – odczyn alergiczny, zaburzenia metabolizmu
białek osocza, zmiany w szpiku kostnym
i chromosomach.
Niedobór związków chromu(III) moŜe mieć wpływ na
rozwój chorób układu krąŜenia i cukrzycy.
Nadmiar – sole chromu(VI) powodują podraŜnienia
śluzówki, skóry, mogą teŜ oddziaływać mutagennie
i kancerogennie.
8
Wartości dopuszczalne
w glebie lub ziemi wg Rozp.
Min. Środ. z dn. 9.09.2002
(w warstwie powierzchn.)
[mg*kg-1]
Grupa
A*
Grupa
B*
Grupa
C*
1
4
15
50
100
600
0,5
2
30
100
300
1000
30
150
600
35
100
300
50
150
500
© Paweł Miśkowiec, ZCHŚ UJ
*
Rozporządzenie definiuje grupy A, B i C jako:
1) grupa A:
a) nieruchomości gruntowe wchodzące w skład obszaru poddanego ochronie na podstawie
przepisów ustawy — Prawo wodne,
b) obszary poddane ochronie na podstawie przepisów o ochronie przyrody; jeŜeli utrzymanie
aktualnego poziomu zanieczyszczenia gruntów nie stwarza zagroŜenia dla zdrowia ludzi lub
środowiska
2) grupa B: grunty zaliczone do uŜytków rolnych z wyłączeniem gruntów pod stawami i gruntów
pod rowami, grunty leśne oraz zadrzewione i zakrzewione, nieuŜytki, a takŜe grunty zabudowane
i zurbanizowane z wyłączeniem terenów przemysłowych, uŜytków kopalnych oraz terenów
komunikacyjnych;
3) grupa C: tereny przemysłowe, uŜytki kopalne, tereny komunikacyjne.
Właściwości gleb a ich podatność na zanieczyszczenie
Bezwzględna zawartość danego metalu cięŜkiego w glebie nie jest jednak wystarczającym
kryterium do oceny tzw. biodostępności danego pierwiastka. Ocena taka przeprowadzona na
uŜytkach rolnych ma niebagatelne znaczenie przy oszacowaniu ilości toksykanta, która moŜe
włączyć się w obieg biogeochemiczny, a tym samym mogącej trafić do konsumenta ostatecznego
jakim jest człowiek. Celem lepszego zrozumienia tematu, naleŜy zapoznać się z kilkoma
podstawowymi pojęciami i definicjami:
Gleba jest złoŜonym, dynamicznym tworem przyrody, odznaczającym się swoistymi cechami
morfologicznymi oraz właściwościami fizycznymi, chemicznymi i biologicznymi, stwarzającym
warunki dla Ŝycia roślin i zwierząt.
Gleba składa się z trzech faz:
stałej – obejmującej cząstki mineralne, organiczne i organiczno-mineralne o róŜnym stopniu
rozdrobnienia, tworzące kompleks sorpcyjny,
ciekłej – tzw. roztwór glebowy, czyli woda, w której są rozpuszczone związki mineralne
i organiczne,
gazowej – tzw. powietrze glebowe, czyli mieszanina gazów, w której stosunki poszczególnych
składników są inne niŜ w powietrzu atmosferycznym.
Faza stała gleby składa się ze składników mineralnych (okruchy skał, minerały, substancje
mineralne) oraz składników organicznych (próchnica i wchodzące w jej skład kwasy humusowe,
resztki roślinne, zwierzęce, organizmy glebowe).
Masa mineralna gleb zawiera ziarna o róŜnej wielkości. RóŜnice w wielkości cząstek oraz proporcje
między nimi wywierają duŜy wpływ na właściwości fizykochemiczne gleby np. wielkość
powierzchni właściwej czy przewiewność. Podział frakcji w zaleŜności od granicznych średnic
ziaren przedstawia się następująco:
frakcja kamieni: ⌀ > 20 mm,
frakcja Ŝwiru: ⌀ 20 – 1 mm,
frakcja piasku: ⌀ 1 – 0,1 mm,
frakcja pyłu: ⌀ 0,1 – 0,02 mm,
frakcja ilasta (części spławialne): ⌀ < 0,02 mm.
9
© Paweł Miśkowiec, ZCHŚ UJ
RównieŜ właściwości kwasowo-zasadowe gleb mają istotny wpływ na przebieg procesów
glebowych, przyswajalność składników odŜywczych, rozwój roślin i mikroorganizmów.
WyróŜniamy:
gleby kwaśne, pH < 6,6, które dzielimy na:
o silnie kwaśne, pH < 4,5;
o kwaśne, pH 4,6 - 5,5;
o lekko kwaśne pH 5,6 - 6,5;
gleby obojętne, pH 6,6 - 7,2;
gleby zasadowe, pH > 7,2.
Formy występowania metali cięŜkich w glebie
Metale cięŜkie są akumulowane w glebie najczęściej w następujących formach:
czynne – bezpośrednio rozpuszczalne w wodzie, występujące w roztworze glebowym
w postaci jonów lub rozpuszczalnych kompleksowych połączeń z ligandami mineralnymi lub
organicznymi;
wymienne – adsorbowane na pozycjach jonowymiennych mineralnych i organicznych
składników kompleksu sorpcyjnego gleby;
specyficznie sorbowane – tworzące silne wiązania koordynacyjne z grupami funkcyjnymi
uwodnionych tlenków Ŝelaza i glinu oraz struktur krzemianowych;
okludowane – (współstrącone) w tlenkach Fe, Al i Mn lub obecne w nich wskutek dyfuzji
wewnątrzkrystalicznej;
związane z substancją organiczną – głównie w formie kompleksów metaloorganicznych.
Kompleksy niskocząsteczkowe charakteryzują się stosunkowo duŜą rozpuszczalnością, natomiast
wielkocząsteczkowe (np. z kwasami huminowymi) są zazwyczaj nierozpuszczalne w wodzie;
chemicznie sorbowane – tworzące wtórne, trudno rozpuszczalne połączenia amorficzne lub
krystaliczne, np. węglany, siarczki, fosforany.
rezydualne – wbudowane w sieci krystaliczne pierwotnych i wtórnych minerałów glebowych
(np. minerałów ilastych); zasadniczo nie są one podatne na działanie nawet silnie agresywnych
czynników, takich jak stęŜone mocne kwasy mineralne.
Formy, w jakich metale cięŜkie występują w glebach, zaleŜą od rodzaju metalu, jego chemicznych
właściwości, pierwotnej postaci w jakiej zostały wprowadzone do gleby, a takŜe od właściwości
samej gleby Miedź i ołów wykazują bardzo silne powinowactwo z substancją organiczną, cynk,
miedź i mangan okludują z tlenkami Ŝelaza, a chrom i nikiel występują zazwyczaj we frakcji
rezydualnej. Kadm i cynk charakteryzują się stosunkowo wysokim udziałem frakcji rozpuszczalnej i
wymiennej, zwłaszcza w glebach kwaśnych.
Czynniki decydujące o rozpuszczalności metali cięŜkich w glebach
Do najwaŜniejszych czynników decydujących o rozpuszczalności (a co za tym idzie,
o fitodostępności) metali cięŜkich w glebach zalicza się zwykle:
pochodzenie i pierwotną formę chemiczną metalu;
odczyn gleby;
pojemność sorpcyjną;
potencjał redoks;
tworzenie połączeń kompleksowych z substancją organiczną.
Głównym czynnikiem decydującym o rozpuszczalności metali cięŜkich w glebie jest odczyn, od
którego zaleŜy m in. połoŜenie równowagi procesów sorpcji i desorpcji kationów wodorowych i
kationów metali. Rozpuszczalność metali cięŜkich warunkowana procesami sorpcji wymiennej jest
niska w zakresie odczynów obojętnych i alkalicznych i rośnie wraz z obniŜaniem wartości pH.
Wzrost zakwaszenia gleby prowadzi zazwyczaj do powolnego rozpuszczania się tlenków Fe, Al
10
© Paweł Miśkowiec, ZCHŚ UJ
i Mn, a nawet minerałów pierwotnych i wtórnych, w wyniku czego następuje uwalnianie z nich
metali cięŜkich. W związku z tym, Ŝe rozpuszczalność metali w głównej mierze zaleŜy od odczynu,
wapnowanie gleb zanieczyszczonych metalami cięŜkimi jest podstawowym zabiegiem
ograniczającym ich mobilność.
Procesy sorpcji i desorpcji kationów oraz sorpcji wymiennej są najwaŜniejszymi mechanizmami
decydującymi o przechodzeniu metali cięŜkich z fazy stałej do roztworu glebowego. Przy ocenie
zagroŜenia związanego z uwalnianiem metali cięŜkich z gleb, a takŜe przy określaniu warunków
ich rolniczego wykorzystania uwzględnia się fakt, Ŝe metale cięŜkie wiązane są silniej w glebach
o większej pojemności sorpcyjnej i cięŜszym składzie granulometrycznym. WaŜnym czynnikiem
decydującym o dostępności metali cięŜkich dla roślin jest teŜ zawartość substancji organicznej w
glebie – zwłaszcza próchnicy glebowej. Wykazuje ona wysoką zdolność sorpcji kationów i ma
bardzo istotny udział w wiązaniu metali cięŜkich z roztworu glebowego. Wzbogacanie kompleksu
sorpcyjnego gleby w substancję organiczną oraz inne dodatki o bardzo silnie rozwiniętym
kompleksie sorpcyjnym, uwaŜane jest, obok wapnowania, za podstawowy zabieg rekultywacyjny na
glebach lekkich zanieczyszczonych metalami cięŜkimi.
Podział gleb wg IUNG
W zaleŜności od omówionych właściwości, gleby mogą być w róŜnym stopniu podatne na
zanieczyszczenia metalami cięŜkimi. Instytut Uprawy NawoŜenia i Gleboznawstwa w Puławach
(IUNG), będący jednostką badawczo-rozwojową, podległą Ministerstwu Rolnictwa i Rozwoju Wsi,
dokonał w związku z tym podziału gleb Polski na trzy grupy. Są to, według wzrastającej odporności
na zanieczyszczenia, gleby grup: AG, BG, CG3 . Podział ten uwzględnia ich:
- odczyn,
- skład granulometryczny (% frakcji < 0,02 mm, tzw. spławialnej), oraz
- zawartość substancji organicznej.
Właściwości te bowiem decydują o dostępności dla roślin metali cięŜkich w glebach. Podział ten
zaprezentowany jest w tabeli 2.
Tabela 2. Podział na grupy gleb mineralnych (wg FS i pH) i organicznych (wg OS)
Odczyn(pH)
<4.5 4.6-5,5
5.6 - 6,5
>6.5
< 10
AG
AG
AG
AG
AG
AG
AG
BG
zawartość
frakcji 10-20
spławialnej FS (%)
20-35
BG
BG
CG
CG
35-55
BG
BG
CG
CG
BG
BG
BG
BG
zawartość
substancji 6-10
*
organicznej OS (%)
>10
CG
CG
CG
CG
*
w glebach organicznych nie uwzględnia się wpływu pH.
Jak wynika z powyŜszej tabeli, w przypadku gleb mineralnych (o zawartości substancji organicznej
< 6%) decydującą rolę odgrywa jej odczyn oraz zawartość frakcji spławialnej. W przypadku gleb
mineralno-organicznych oraz organicznych, o jej przypisaniu do danej grupy decyduje jedynie
zawartość substancji organicznej.
Graniczne zawartości metali cięŜkich w glebach róŜnych grup i o róŜnych stopniach
zanieczyszczenia zaprezentowane są w tabeli 3.
3
Uwaga – proszę nie mylić grup gleb AG, BG i CG wprowadzonych przez IUNG z grupami gleb A, B i C, które
definiuje Minister Środowiska. To są dwa zupełnie inne i niezwiązane ze sobą podziały.
11
© Paweł Miśkowiec, ZCHŚ UJ
Tabela 3. Graniczne zawartości metali cięŜkich w glebach o róŜnych stopniach zanieczyszczenia wg
IUNG [mg*kg-1].
Stopień zanieczyszczenia gleb
Grupa
Metal
gleby 0
I
II
III
IV
V
AG
0,3
1
2
3
5
>5
BG
0,5
1,5
3
5
10
>10
Cd
CG
1
3
5
10
20
>20
AG
20
70
100
500
2500
>2500
BG
40
100
250
1000
5000
>5000
Pb
CG
60
150
500
2000
7000
>7000
AG
50
100
200
700
1500
>1500
BG
70
150
300
1000
3000
>3000
Zn
CG
100
250
500
2000
5000
>5000
AG
10
30
50
80
300
>300
BG
20
50
80
100
500
>500
Cu
CG
25
70
100
150
750
>750
AG
10
30
50
100
400
>400
BG
25
50
75
150
600
>600
Ni
CG
50
75
100
300
1000
>1000
AG
20
40
80
150
300
>300
BG
30
60
150
300
500
>500
Cr
CG
50
80
200
500
1000
>1000
IUNG zaleca rolnicze uŜytkowanie gleb proponując sześciostopniową klasyfikację w zaleŜności
od stopnia zanieczyszczenia metalami cięŜkimi:
Stopień 0 - gleby nie zanieczyszczone o naturalnych zawartościach metali śladowych. Gleby te
mogą być przeznaczone pod wszystkie uprawy ogrodnicze i rolnicze, zgodnie z zasadami
racjonalnego wykorzystania rolniczej przestrzeni produkcyjnej.
Stopień I - obejmuje gleby o podwyŜszonej zawartości metali. Gleby te mogą być przeznaczone
pod wszystkie uprawy polowe, z ograniczeniem warzyw przeznaczonych dla dzieci.
Stopień II - gleby słabo zanieczyszczone. Na glebach takich zachodzi juŜ obawa chemicznego
zanieczyszczenia roślin. Wykluczyć więc naleŜy przede wszystkim niektóre uprawy ogrodnicze, jak
np. sałata, szpinak, kalafior. Dozwolona jest uprawa roślin zboŜowych, okopowych i pastewnych.
Stopień III - gleby średnio zanieczyszczone. Wszystkie uprawy na takich glebach naraŜone są na
skaŜenie. Dopuszczalna jest uprawa roślin zboŜowych, okopowych i pastewnych pod warunkiem
okresowej kontroli poziomu metali w konsumpcyjnych częściach roślin. Zalecane są uprawy roślin
przemysłowych i traw nasiennych.
Stopień IV - gleby silnie zanieczyszczone. Gleby takie (szczególnie gleby lekkie) powinny być
wyłączone z produkcji rolniczej oraz zadarnione lub zadrzewione. Na glebach lepszych moŜna
uprawiać rośliny przemysłowe (len, konopie, wiklina). Dopuszcza się produkcję materiału
siewnego zbóŜ i traw, a takŜe ziemniaków dla przemysłu spirytusowego (na spirytus jako dodatek
do paliwa) i rzepaku na olej techniczny. Zaleca się zabiegi rekultywacyjne, a głównie wapnowanie
i wprowadzanie substancji organicznej.
Stopień V - gleby bardzo silnie zanieczyszczone. Gleby o takim stopniu zanieczyszczenia naleŜy
wyłączyć z produkcji rolniczej i poddać zabiegom rekultywacyjnym. MoŜna uprawiać (na glebach
przydatnych) len, konopie oraz rzepak (na olej techniczny), a w dolinach rzek – wiklinę.
12
© Paweł Miśkowiec, ZCHŚ UJ
Ćwiczenie
Oznaczanie zawartości metali cięŜkich w glebie metodą atomowej spektrometrii absorpcyjnej
(ASA)
Cel ćwiczenia
Celem ćwiczenia jest oznaczenie całkowitej zawartości wybranych metali cięŜkich w glebach oraz
zawartości form najbardziej mobilnych, ustalenie czy przekroczone są wartości dopuszczalne stęŜeń
określone przez Ministra Środowiska, jak równieŜ określenie stopnia zanieczyszczenia według
kryteriów IUNG oraz ich przydatności rolniczej.
Przyrządy, materiały, odczynniki
Kwas azotowy(V) stęŜony cz.d.a.
Kwas azotowy(V) 2%
Perhydrol cz.d.a.
1 M octan amonu (pH 7)
Roztwory wzorcowe Cd, Ni, Pb, Zn, Cu
Absorpcyjny spektrometr atomowy AAnalyst 300 firmy Perkin Elmer,
Mineralizator mikrofalowy Magnum II firmy Ertec,
Wytrząsarka, wirówka
Lejki szklane,
Kolby miarowe,
Probówki typu Falcon
Filtry
Wykonanie ćwiczenia:
Oznaczenie całkowitej zawartości metali
OdwaŜyć ok. 1g powietrznie suchej gleby, przesianej wcześniej przez sito o średnicy oczek 2mm.
Zanotować masę gleby z dokładnością do 4 cyfr dziesiętnych. Próbkę glebową przenieść ilościowo
do naczynia teflonowego i zadać 7 cm3 stęŜonego kwasu azotowego(V) oraz 3 cm3 roztworu
perhydrolu. Następnie próbkę zmineralizować dwustopniowo według programu z tabeli 4.
Tabela 4. Warunki pracy mineralizatora mikrofalowego MAGNUM II podczas procesu
mineralizacji próbek gleby.
Parametr
Czas ogrzewania [min]
Ciśnienie max [bar]
Ciśnienie min [bar]
Temperatura max [°C]
Temperatura min [°C]
Moc [%]
Czas chłodzenia po mineralizacji [min]
ETAP 1
5
33
30
260
255
60
ETAP 2
20
45
43
260
255
80
10
Po zakończeniu mineralizacji przesączyć zawartość naczynka teflonowego do kolby miarowej
o objętości 25 cm3 i uzupełnić do kreski.
Z wyjściowych roztworów wzorcowych zawierających metale cięŜkie sporządzić przez
rozcieńczenie w 2% roztworze HNO3 roztwory o stęŜeniach obejmujących zakres liniowej
zaleŜności absorbancji od stęŜenia.
Zgodnie z instrukcją obsługi spektrometru ASA wyznaczyć krzywe kalibracyjne dla
poszczególnych pierwiastków, a następnie zmierzyć stęŜenie metali w analizowanych roztworach.
Zanotować niepewność oznaczenia.
13
© Paweł Miśkowiec, ZCHŚ UJ
Uwaga 1: roztwory nie mogą zawierać zawiesin.
Uwaga 2: jeŜeli stęŜenie metalu w badanej próbce przekracza zakres krzywej kalibracyjnej,
konieczne jest rozcieńczenie próbki.
Oznaczenie form czynnej i wymiennej metali w glebie
OdwaŜyć w probówce typu Falcon ok. 2 g powietrznie suchej gleby, przesianej wcześniej przez sito
o średnicy oczek 2 mm. Zanotować masę gleby z dokładnością do 4 cyfr dziesiętnych. Próbkę
zadać 20 cm3 1 M roztworu octanu amonu (pipetą). Wytrząsać przez 1 h. Po zakończeniu
wytrząsania odwirować przez 10 min, a następnie przenieść ok. 15 cm3 klarownego roztworu znad
osadu do pojemnika pomiarowego.
Z wyjściowych roztworów wzorcowych zawierających metale cięŜkie sporządzić przez
rozcieńczenie w 1M roztworze CH3COONH3 roztwory o stęŜeniach obejmujących zakres liniowej
zaleŜności absorbancji od stęŜenia.
Opracowanie wyników
Na podstawie uzyskanych wyników:
- obliczyć zawartość badanych metali w glebie w mg/kg, najlepiej poprzez wyprowadzenie
zaleŜności zawartości metalu w glebie „s” od odczytanego ze spektrometru stęŜenia „c”, masy
próbki glebowej „m” oraz objętości kolby miarowej „v1” (lub w przypadku octanu amonu objętości
uŜytej pipety „v2”).
- obliczyć niepewność uzyskanego wyniku, najlepiej metodą róŜniczki zupełnej, mając, oprócz
danych powyŜszych, następujące wartości niepewności:
∆ masy próbki glebowej = 0,0001 g,
∆ objętości kolby miarowej 25 cm3 = 0,04 cm3,
∆ objętości pipety 20 cm3 = 0,04 cm3,
∆ stęŜenia metalu w roztworze – odczytana z urządzenia.
Przy ewentualnym rozcieńczeniu:
∆ rozcieńczenia 10 krotnego = 0,05,
∆ rozcieńczenia 100 krotnego = 2,1.
Przy opracowywaniu wyników proszę pamiętać, Ŝe:
- oszacowane niepewności zaokrąglamy zawsze w górę,
- obliczenia wykonujemy zawsze z większą liczbą cyfr, niŜ chcemy podać wynik. Zaokrągleń
dokonujemy dopiero po zakończeniu obliczeń,
- niepewności zaokrąglane są do maksymalnie dwóch cyfr znaczących. Wartość niepewności naleŜy
zaokrąglić do jednej cyfry znaczącej, jeŜeli nie zmieni to wartości niepewności o więcej niŜ 10%,
- ostatnia cyfra znacząca w kaŜdym wyniku pomiaru powinna stać na tym samym miejscu
dziesiętnym, co ostatnia cyfra niepewności pomiarowej.
Mając dany wynik oraz jego niepewność naleŜy:
Dla zawartości całkowitej metalu
- sprawdzić czy w badanej glebie nie przekroczone zostały wartości dopuszczalne stęŜeń
wyszczególnionych w Rozporządzeniu Ministra Środowiska,
- ocenić klasę zanieczyszczenia gleby wg klasyfikacji IUNG,
- podać jej przydatność do celów rolniczych.
Dla zawartości form fitodostępnych
- obliczyć jaki procent z zawartości całkowitej stanowią sumarycznie formy czynne i wymienne
Z całości uzyskanych wyników wyciągnąć odpowiednie wnioski.
Wszystkie obliczenia proszę umieścić w sprawozdaniu.
14
© Paweł Miśkowiec, ZCHŚ UJ
Zakres materiału naukowego
Literatura podstawowa:
1. Szczepaniak W.: Metody instrumentalne w analizie chemicznej, PWN, Warszawa 2008,
str. 143-166.
2. Cygański A.: Metody spektroskopowe w chemii analitycznej, Wydawnictwa NaukowoTechniczne, Warszawa 2009, str. 32-45, 128-137.
3. śernicki W., Borkowska-Burnecka J., Bulska E., Szmyd E.: Metody analitycznej
spektrometrii atomowej - teoria i praktyka, Wydawnictwo Malamut, Warszawa 2010, str. 70105.
Literatura uzupełniająca:
1. Kabata-Pendias A., Piotrowska M.: Podstawy oceny chemicznego zanieczyszczenia gleb.
Metale cięŜkie, siarka i WWA. Biblioteka Monitoringu Środowiska, PIOŚ, IUNG, Warszawa,
1995, str. 16-19.
2. Kabata-Pendias A., Pendias H.: Biogeochemia pierwiastków śladowych, PWN, Warszawa
1999. Rozdziały z informacjami o poszczególnych metalach cięŜkich.
3. Rosada J., J. Grzesiak, P. Grzesiak, G. Schroeder, I. Ratajczak, I. Rissmann. 2004. Oznaczanie
metali cięŜkich w glebach objętych emisjami przemysłowymi metoda ekstrakcji
sekwencyjnej, Chemiczne aspekty badań środowiska red.: G. Schroeder, 2, 143-162.
4. Rozporządzenie Ministra Środowiska z dnia 9 września 2002 r. w sprawie standardów jakości
gleby oraz standardów jakości ziemi. Dz.U. 2002 nr 165 poz. 1359,
http://isap.sejm.gov.pl/DetailsServlet?id=WDU20021651359
15