Prace poglądowe
Dent. Med. Probl. 2012, 49, 1, 95–102
ISSN 1644-387X
© Copyright by Wroclaw Medical University
and Polish Dental Society
Elżbieta Dembowska, Maria Wiernicka-Menkiszak, Renata Samulak-Zielińska
Uogólnione agresywne zapalenie przyzębia
– diagnostyka, występowanie, etiopatogeneza
Generalized Aggressive Periodontitis – Diagnostic, Epidemiology,
Etiopathogenesis
Zakład Periodontologii Pomorskiego Uniwersytetu Medycznego w Szczecinie
Streszczenie
Uogólnione agresywne zapalenia przyzębia (GAgP – generalized aggressive periodontitis) to jednostka chorobowa,
dla której nomenklatura i kryteria diagnostyczne zostały ustalone w 1999 r. w USA podczas Międzynarodowych
Warsztatów Periodontologicznych. Po upływie ponad 10 lat wiadomo, że jest ono skutkiem nieprawidłowości
odpowiedzi immunologicznej organizmu gospodarza na czynnik infekcyjny. Udokumentowane do dzisiaj nieprawidłowości dotyczą zmniejszenia możliwości fagocytozy drobnoustrojów przez granulocyty obojętnochłonne
oraz zmniejszonego wydzielania przez nie przeciwbakteryjnego białka hCAP-18/LL-37. Oczywiste jest zachwianie
równowagi między cytokinami wydzielanymi przez limfocyty i monocyty/makrofagi, a w uogólnionym agresywnym zapaleniu przyzębia w porównaniu z zapaleniami przewlekłymi stwierdza się zmniejszone wydzielanie przeciwzapalnej IL-10 i zmniejszone stężenie przeciwciał klasy IgG przeciwko Aggregatibacter actinomycetemcomitans
i Porphyromonas gingivalis z jednocześnie zwiększoną liczbą wymienionych periopatogenów. Wpływ genów na
powstawanie agresywnych zapaleń przyzębia jest pewny. Nie do końca wiadomo jednak, w jaki sposób regulują
one ekspresję chorób. Diagnostyka różnicowa uogólnionego agresywnego zapalenia przyzębia i zaawansowanego
uogólnionego przewlekłego zapalenia przyzębia nie jest jednak jednoznaczna. Wydaje się, że pacjenci sklasyfikowani obecnie jako osoby chorujące na GAgP nie stanowią jednostki diagnostycznie jednolitej, a obecne wyniki badań
sugerują konieczność kolejnej zmiany w klasyfikacji chorób przyzębia (Dent. Med. Probl. 2012, 49, 1, 95–102).
Słowa kluczowe: uogólnione agresywne zapalenie przyzębia, epidemiologia, histologia, etiopatogeneza.
Abstract
Generalized aggressive periodontitis (GAgP) is a disease for which the nomenclature and diagnostic criteria were
established in 1999 in the USA during the International Workshop for Periodontics. After more than 10 years,
we know that GAgP is the result of irregularities in the host organism’s immune response to an infectious agent.
Documented to date irregularities are: the reduce of possibility of the phagocytosis of microorganisms by neutrophils and reduced secretion of antimicrobial proteins by hCAP-18/LL-37. Imbalance between the cytokines
produced by lymphocytes and monocytes/macrophages is obvious and in generalized aggressive periodontitis,
compared to chronic inflammatory states, there is reduced secretion of anti-inflammatory IL-10 and decreased
levels of IgG antibodies to Aggregatibacter actinomycetemcomitans and Porphyromonas gingivalis with in the
same time increased number of those bacteria. Influence of genes on the formation of aggressive periodontitis is
uncertain. However, we do not understand how genes regulate the expression of the disease. The differential diagnosis of generalized aggressive periodontitis and advanced generalized chronic periodontitis is not clear. It seems
that in patients currently classified as those suffering from GAgP the disease is not a single diagnostic entity and the
current results in research suggest the need for another change in the classification of periodontal diseases (Dent.
Med. Probl. 2012, 49, 1, 95–102).
Key words: generalized aggressive periodontitis, epidemiology, histology, etiopathogenesis.
96
E. Dembowska, M. Wiernicka-Menkiszak, R. Samulak-Zielińska
Klasyfikacja i nazewnictwo chorób przyzębia zmieniają się z czasem. Osiągnięcia naukowe
z zakresu badań laboratoryjnych w takich dziedzinach, jak mikrobiologia, immunologia i genetyka,
wymuszają nowe spojrzenie na wiele jednostek
chorobowych, w tym na tak niejednolitą jednostkę
diagnostyczną, jaką jest zapalenie przyzębia (periodontitis).
W 1999 r. w Illinois w USA odbyły się Międzynarodowe Warsztaty Periodontologiczne, podczas których zatwierdzono obowiązującą w Polsce
i na świecie bardzo rozbudowaną klasyfikację stanów chorobowych związanych z przyzębiem. Dla
grupy zapaleń przyzębia cechujących się szybkim,
a niekiedy gwałtownym przebiegiem klasyfikowanych do 1999 r. jako early-onset periodontitis przyjęto nowe kryteria diagnostyczne i podzielono na
różne jednostki chorobowe. Jedną z nich jest bardzo trudne do rozpoznania i leczenia uogólnione agresywne zapalenie przyzębia (GAgP – generalized aggressive periodontitis). Kryteria diagnostyczne dla tej jednostki chorobowej tak bardzo
zmieniły się w porównaniu z wcześniejszymi podziałami chorób przyzębia, że nie można dokonać
prostego porównania badań sprzed i po 2000 r.
Nowy podział i nowe kryteria diagnostyki
chorób przyzębia w dalszym ciągu nie są wystarczająco jasne i wymagają utworzenia dla wprowadzonej klasyfikacji nowej bazy danych uwzględniającej odmienne kryteria stanu przyzębia, szczególnie dla uogólnionego agresywnego zapalenia
przyzębia.
Celem tego opracowania jest zebranie obecnej wiedzy na temat występowania, etiopatogenezy i diagnostyki klinicznej uogólnionego agresywnego zapalenia przyzębia.
Diagnostyka
Zgodnie z przyjętymi w 1999 r. kryteriami
diagnostycznymi określenie stanu przyzębia jako
agresywne zapalenia przyzębia może nastąpić po
spełnieniu niżej wymienionych warunków [1].
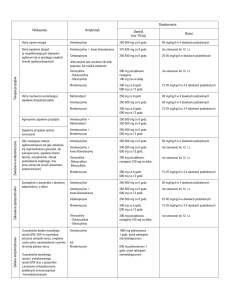
Objawy pierwszorzędowe:
– pacjenci oprócz zapalenia przyzębia nie mają
innych chorób,
– dochodzi do szybkiej utraty przyczepu łącznotkankowego i szybkiej utraty kości wyrostka zębodołowego szczęki i części zębodołowej żuchwy,
– schorzenie cechuje się częstym występowaniem rodzinnym.
Objawy drugorzędowe:
– ilość złogów bakteryjnych nie ma żadnego
związku ze stopniem zaawansowania choroby,
– obecność dużej liczby takich drobnoustrojów, jak Aggregatibacter actinomycetemcomitans
(Aa) i Porphyromonas gingivalis (Pg) w poddziąsłowym biofilmie,
– nieprawidłowa fagocytoza granulocytów
obojętnochłonnych,
– prawdopodobne jest występowanie nadmiernej aktywności makrofagów, które wykazują
wzmożoną sekrecję niektórych mediatorów zapalenia, takich jak prostaglandyna 2 (PGE2) oraz interleukina-1 beta (IL-1β),
– postępująca utrata przyczepu łącznotkankowego może się samoistnie zatrzymać.
Aby rozpoznać uogólnione agresywne zapalenie przyzębia, należy przyjąć dodatkowe kryteria,
które zgodnie z postanowieniami Międzynarodowych Warsztatów Periodontologicznych z 1999 r.
są następujące [1]:
– początek choroby najczęściej około 30. r.ż.,
ale pacjenci mogą być również starsi,
– słaba reakcja immunologiczna na czynniki
infekcyjne i małe stężenia przeciwciał przeciwko
bakteriom patogennym obecnych w przyzębiu,
– skokowa utrata przyczepu łącznotkankowego i kości,
– uogólniona osteoliza kości w przestrzeniach
międzyzębowych obejmująca przynajmniej trzy
zęby stałe z wykluczeniem centralnych siekaczy
szczęki oraz pierwszych zębów trzonowych,
– w odróżnieniu od umiejscowionego agresywnego zapalenia przyzębia (LAgP), w którym
ilość złogów bakteryjnych nie ma związku z zaawansowaniem choroby, w uogólnionym agresywnym zapaleniu przyzębia (GAgP) zależność ta może zaistnieć.
Epidemiologia
Podanie danych epidemiologicznych dotyczących częstości występowania agresywnego zapalenia przyzębia nie jest łatwe. Demmer i Papanaou [2] dokonali przeglądu piśmiennictwa znajdującego się w bazie bibliograficznej PubMed
i zawierającego hasło „agresywne zapalenia przyzębia” (aggressive periodontitis) ogłoszonego drukiem do lipca 2009 r. i znaleźli na ten temat 1650–
–3300 publikacji, przy czym termin ten był stosowany już w pracach z 1948 r.
Po dokonaniu wstępnego przeglądu publikacji
do analizy zakwalifikowano wszystkie prace anglojęzyczne opublikowane po 1 stycznia 2000 r., które zawierały w słowach kluczowych słowa „periodontal” lub „periodontitis” i dotyczyły badań przeprowadzonych na ludziach (289 prac). Większość
z nich zawierała dane, które nie spełniały kryteriów diagnostycznych przyjętych do rozpoznania
GAgP w 1999 r. podczas Warsztatów Periodontologicznych. W niektórych prowadzono obserwa-
Uogólnione agresywne zapalenie przyzębia
cje na pacjentach ze schorzeniami ogólnoustrojowymi, np. cukrzycą. Jeszcze inne uwzględniały
jedynie pacjentów zgłaszających się z już istniejącą chorobą przyzębia lub były wiernym powtórzeniem wcześniejszych badań i nie mogą stanowić danych epidemiologicznych. Po odrzuceniu
tych prac do analizy pozostało jedynie 21 publikacji, z których tylko jedna spełniała wszystkie kryteria dotyczące częstości występowania uogólnionego agresywnego zapalenia przyzębia (generalized aggressive periodontitis). Badanie to zostało
przeprowadzone przez Levina et al. [3] i dotyczyło
populacji młodych Izraelitów w wieku 18–30 lat,
kobiet i mężczyzn odbywających służbę w izraelskim wojsku. Częstość występowania uogólnionego agresywnego zapalenia przyzębia w tej populacji wyniosła 2%.
Polskie badania epidemiologiczne przeprowadzone w 2011 r. przez ośrodki akademickie w Warszawie, Białymstoku, Szczecinie, Lublinie i Wrocławiu oraz prywatną praktykę w Kielcach na losowo wybranych osobach zamieszkujących duże
polskie miasta opisują częstość, stopień zaawansowania i potrzeby lecznicze na podstawie wskaźnika CPITN u osób w wieku 35–44 lata [4]. Z badań
tych wynika, że odsetek osób z zaawansowanym
zapaleniem przyzębia (kieszonki o głębokości 5,5
mm i więcej) wyniósł aż 16,5%. Głębokie kieszonki przyzębne występowały statystycznie częściej
u mężczyzn (24,2% badanych) niż u kobiet (13,6%
badanych). W grupie pacjentów z zapaleniami
przyzębia 95,9% stanowiły osoby z przewlekłym
zapaleniem przyzębia, 2,8% z agresywnym przyzębia, a u 1,4% zapalenie przyzębia było związane
z chorobami ogólnoustrojowymi.
Immunohistopatologia
W przeciwieństwie do zapaleń dziąseł nie ma
takich badań histologicznych, które scharakteryzowałyby postęp uogólnionego agresywnego zapalenia przyzębia. Dzieje się tak z kilku powodów;
po pierwsze, liczba osób, których dotyczy to schorzenie jest niewielka, po drugie, z powodu ciągłych zmian w nomenklaturze i kryteriach diagnostycznych. W różnych okresach do agresywnych zaplenia przyzębia zaliczano: cementopatię
(Gottlieb 1926), zanik ostry (Parma 1941), periodonthopathia dystrophica (ARPA-Internationale
1955), paradontozę (ARPA-Internationale 1967),
głębokie brzeżne zapalenie przyzębia – periodontitis marginalis profunda (Page, Schroeder 1982),
przedpokwitaniowe zapalenie przyzębia – prepubertal periodontitis (Page 1983), szybko postępujące zapalenie przyzębia – rapidly progressive periodontitis (Schroeder 1983), młodzieńcze zapale-
97
nie przyzębia – juvenile periodontitis, oporne na
leczenie zapalenie przyzębia (refractory periodontitis), wczesne periodontitis (early-onset periodontitis), zlokalizowane agresywne zapalenie przyzębia – localized aggressive periodontitis, uogólnione agresywne zapalenie przyzębia – generalized
aggressive periodontitis (1999 International Workshop for a Classification of Periodontal Diseases
and Conditions). Trzecim powodem jest sam przebieg uogólnionego agresywnego zapalenia przyzębia. Jego początek może być klinicznie identyczny jak w zaawansowanym przewlekłym zapaleniu przyzębia. Dotyczy to zwłaszcza pacjentów po
30. r.ż.
Dostępne publikacje na temat histologicznego
stanu tkanek przyzębia pochodzą z lat wcześniejszych i dotyczą starszych podziałów, co wydaje się
nie umniejszać ich wartości, ponieważ kolejne doniesienia na ten temat opierają się właśnie na tych
badaniach [5–7]. Stwierdzono wówczas, że pod
względem histologicznym obraz tkanek w przewlekłym zapaleniu przyzębia i we wczesnych postaciach agresywnych chorób przyzębia (earlyonset periodontitis) jest podobny. Miejscową reakcją gospodarza na drobnoustroje biofilmu jest
gromadzenie się wzdłuż granicy nabłonka i tkanki łącznej granulocytów obojętnochłonnych, które
tworzą barierę między płytką bakteryjną i tkankami przyzębia. Komórki te migrują także do płynu kieszonki, a u osób z agresywnym zapaleniem
przyzębia ściśle pokrywają powierzchnię korzenia. W zaawansowanych postaciach zapaleń przyzębia tkanka łączna jest nacieczona głównie komórkami plazmatycznymi, które są stymulowane
za pomocą limfocytów CD4. Skutkiem tego zjawiska jest nagromadzenie w tkance łącznej dużej
ilości immunoglobulin klasy IgG. Obserwuje się
także zniszczenie istoty międzykomórkowej tkanek i miejscową destrukcję nabłonka kieszonki
i tkanki łącznej. Jest to skutek uwalniania enzymów lizosomalnych jako rezultat nieudanej fagocytozy neutrofilów [8–12]. Nowsze doniesienia na
ten temat to praca Artese et al. [13]. Zbadali oni
liczbę CD4 (limfocyty pomocnicze), CD8 (limfocyty cytotoksyczne/supresorowe), CD20 (komórki plazmatyczne) i CD68 (makrofagi) w tkankach
dziąsła u osób z przewlekłym (CP) i agresywnym
zapaleniem przyzębia (AgP). U osób z CP zaobserwowano więcej CD4 i CD8. U osób z AgP
wszystkie komórki CD4, CD8, CD20, CD68 występowały w większej liczbie, ale w porównaniu
z przewlekłym zapaleniem przyzębia różnicę statystycznie istotną zanotowano jedynie w liczbie
CD20. Naciek z komórek plazmatycznych w tkankach u pacjentów z agresywnym zapaleniem przyzębia był istotnie większy w porównaniu z pacjentami z przewlekłym zapaleniem przyzębia.
98
E. Dembowska, M. Wiernicka-Menkiszak, R. Samulak-Zielińska
Etiopatogeneza
Wpływ bakterii na powstawanie i rozwój chorób przyzębia jest niezaprzeczalny. Każda dyskusja
na temat jakości i liczby drobnoustrojów w przyzębiu musi być jednak oparta na założeniu, że wykaz mikrobiologiczny drobnoustrojów powodujących schorzenia w obrębie przyzębia jest jeszcze niekompletny. Próby przypisania konkretnych
gatunków bakterii do określonego typu zapalenia przyzębia nie powiodły się. Skład mikrobiologiczny kieszonek przyzębnych jest uzależniony
od stopnia zaawansowania choroby i głębokości
kieszonek przyzębnych, w których drobnoustroje mogą współistnieć jedynie w ściśle określonych
kompleksach bakteryjnych [13]. W kieszonkach
o głębokości przekraczającej 4 mm stwierdza się
obecność drobnoustrojów zaliczanych przez Socransky’ego do tzw kompleksów: czerwonego i pomarańczowego zawierających bakterie z rodzaju
Porphyromonas gingivalis (Pg), Tannerella forsythia (Tf) oraz Treponema denticola (Td) oraz Fusobacterium nucleatum (Fn), Prevotella intermedia
(Pi), Parvimonas micra (Pm) (dawniej Peptostreptococcus micros, Micromonas micros). Często spotykanym drobnoustrojem jest również Aggregatibacter actinomycetemcomitans (Aa) [15–20].
Porównanie składu mikrobiogicznego poddziąsłowej flory bakteryjnej u osób z przewlekłym
i agresywnym uogólnionym zapaleniem przyzębia
nie wykazuje znaczących różnic w częstości występowania w biofilmie poddziąsłowym takich bakterii, jak P. gingivalis, P. micra, T. forsytha, A. actinomycetemcomitans, P. intermedia, Capnocytophaga ochracea, Fusobacterium spp., F. nucleatum.
Co więcej, Pg. ingivalis, T. forsythia, C. rectus,
A.actinomycetemcommitans są stwierdzane także
u osób ze zdrowym przyzębiem [21–25]. W populacji Chińczyków wykryto Porphyromonas gingivalis u 76% osób z przewlekłym zapaleniem przyzębia i u 100% pacjentów z uogólnionym agresywnym zapaleniem przyzębia, P. micra zarówno
w CP, jak i GAgP w około 4%. Jedyną istotną statystycznie różnicę wykazano w częstości występowania C. rectus – 23% w CP i 50% w GAgP [21].
W populacji Kolumbijczykow P. gingivalis była
obecna u około 76% osób z CP i u 73% z GAgP,
T. forsythia odpowiednio u 64% i 54%, C. rectus 38% i 32%, A. actinomycetemcomitans w 17%
i 27%, enterococci w 30% i 28% [22]. Badania przeprowadzone przez Riepa et al. [23]. wykazały jedynie większą częstość występowania w kieszonkach
przyzębnych u osób z GAgP Treponema lecithinolyticum, a badania Faveriego [24] częstsze występowanie Selenomonas sp. i Streptococcus sp. w kieszonkach głębszych niż 7 mm. Prace te należy jednak uznać za pilotażowe [25]. Niektórzy badacze
podnoszą problem zakażeń innymi drobnoustrojami, do których należą zakażenia bakteryjne (np.
bakterią Filifactor alocis [26]), wirusowe (wirus
opryszczki zwykłej, HSV-1, wirus Epsteina-Barr
EBV-1, cytomegalowirus HCM) [19], czy grzybami
[27]. Choć te drobnoustroje są wykrywane w głębokich kieszonkach przyzębnych u osób z uogólnionym agresywnym zapaleniem przyzębia, ich
rola w tej patologii nie jest do końca wyjaśniona.
Brak skuteczności leczenia agresywnych form
zapaleń przyzębia przez eliminację płytki bakteryjnej może być częściowo wyjaśniony penetracją
niektórych bakterii poddziąsłowej płytki bakteryjnej do tkanek przyzębia (P. gingivalis, A. actinomycetemcomitans), gdzie stanowią rezerwuar flory bakteryjnej podtrzymującej stan zapalny. Fakt
ten jednak nie tłumaczy do końca dużej dynamiki procesów zapalnych przebiegających w uogólnionym agresywnym zapaleniu przyzębia. Skłoniło to do poszukiwania współistniejących przyczyn szybkiego postępu GAgP. Uwaga skupiła się
obecnie na wyjaśnieniu mechanizmów odpornościowych, zwłaszcza z zakresu wrodzonej, miejscowej i ogólnej odpowiedzi immunologicznej, która w tej jednostce jest niewydolna w stosunku do
obecnej w jamie ustnej flory bakteryjnej. Jej zaburzenie jest uważane dzisiaj za główny czynnik
patogenetyczny, uniemożliwiający eliminację namnożonych patogennych szczepów bakteryjnych.
Eick et al. [28] wykazali, że neutrofile obserwowane w płynie dziąsłowym osób GAgP w porównaniu z neutrofilami u osób ze zdrowym przyzębiem zawierały we wnętrzu zdecydowanie mniej
sfagocytowanych bakterii Porphyromonas gingivalis i obu szczepów A. actinomycetemcomitans.
Uszkodzenie czynności neutrofilów opisali także Türkoğlu et al. [29]. Wykazano, że mimo nagromadzenia granulocytów obojętnochłonnych
w tkankach objętych zapaleniem, w uogólnionym
agresywnym zapaleniu przyzębia występuje miejscowy niedobór wydzielanego przez neutrofile
przeciwbakteryjnego białka hCAP-18/LL-37.
Przyczyn zaburzeń fagocytozy upatruje się
w zmienionych receptorach błon komórkowych
makrofagów, mikrofagów i komórek tucznych.
Dotyczy to zwłaszcza tzw. Fc-receptorów, które
łączą się z kompleksem IgG–drobnoustrój i wywołują proces fagocytozy. Metaanaliza 17 badań [30] przeprowadzonych łącznie na 1685 pacjentach rasy kaukaskiej i azjatyckiej z zapaleniami przyzębia i 1570 ze zdrowym przyzębiem
wykazała słaby związek między polimorfizmem
genu dla Fc-gamma RIIA H131R a uogólnionym
agresywnym zapaleniem przyzębia u rasy azjatyckiej. Związek polimorfizmu genów dla Fc-gamma
RIIIA F158V w uogólnionym agresywnym zapaleniu przyzębia nie istnieje, a polimorfizm genów
Uogólnione agresywne zapalenie przyzębia
dla Fc-gamma RIIIB NA1/NA2 był w równym
stopniu związany z przewlekłym i agresywnym
zapaleniem przyzębia zarówno u rasy azjatyckiej,
jak i kaukaskiej.
Zainteresowanie budzą także tzw. receptory
wrodzonej odporności immunologicznej (TLR –
toll-like receptor). Za ich pośrednictwem są rozpoznawane wzorce molekularne odziedziczone
w rozwoju filogenetycznym i związane z identyfikowaniem patogenów obecnych w naszym środowisku od dawna. Obecne są na powierzchni
błony komórkowej albo w przestrzeni wewnątrzkomórkowej monocytów/makrofagów, komórek
dendrytycznych, komórek tucznych, a także na
powierzchni komórek nabłonków wyścielających
drogi moczowe, oddechowe, przewód pokarmowy, rogówki, dziąsła, na fibroblastach i komórkach śródbłonka, co skutkuje wydzielaniem cytokin. Nie ma jednak wystarczających dowodów
na silne powiązania TLR z uogólnionym agresywnym zapaleniem przyzębia [31].
Teles et al. [32] zaobserwowali, że różne profile
poddziąsłowego biofilmu są związane z odmiennymi wzorcami ekspresji cytokin w płynie dziąsłowym. W tkankach objętych zapaleniem przyzębia klinicznie rozpoznanym jako agresywne
stwierdzili większe stężenie IL-1 beta, większe stężenie czynnika stymulującego neutrofile oraz makrofagi (GM-CSF – granulocyte-macrophage colony-stimulating factor) oraz zaburzony stosunek
IL-1beta/IL-10 w płynie dziąsłowym. Ten ostatni fakt przemawia za brakiem równowagi między
cytokinami pro-(IL-1 beta) i antyzapalnymi (IL10) w uogólnionym agresywnym zapaleniu przyzębia.
Do podobnych wniosków doszli Casarin et al.
[17]. Porównali grupę pacjentów z uogólnionym
agresywnym zapaleniem przyzębia i uogólnionym
przewlekłym zapaleniem przyzębia pod względem
liczby patogenów A.a. i P.g. oraz reakcji immunologicznej wyrażającej się większym stężeniem niektórych cytokin i IgG. W biofilmie pobranym od
pacjentów z GAgP stwierdzono istotnie większą
liczbę drobnoustrojów z rodzaju A.a. i P.g. w porównaniu z biofilmem pacjentów z CP. Ale stężenie przeciwciał klasy IgG w płynie kieszonki przeciwko tym patogenom było u pacjentów z GAgP
istotnie mniejsze. W płynie kieszonki pacjentów
z GAgP zanotowano także zmniejszone stężenie
działającej przeciwzapalnie IL-10.
Rescal et al. [33]. dokonali pomiarów stężenia
IL-1 beta, IL-2, IL-4, IL-8, IFN-gamma i aktywności elastazy. Analiza osiągniętych wyników nie wykazała istotnych statystycznie różnic w mierzonych
kryteriach immunologicznych i mikrobiologicznych u osób z uogólnionym przewlekłym i uogólnionym agresywnym zapaleniem przyzębia.
99
Wyniki badań nad interleukiną pierwszą i genami ją kodującymi w aspekcie czynnika ryzyka
uogólnionego agresywnego zapalenia przyzębia są
sprzeczne. Choć Baradan-Rahimi wykazał, że u Irańczyków stężenie IL-1 u osób z zapaleniami przyzębia
jest zdecydowanie większe niż u osób ze zdrowym
przyzębiem [34], to obecnie nie ma wystarczających
dowodów na związek zmienności genów kodujących
IL-1A i IL-1B i IL-1RN właśnie z agresywnymi zapaleniami przyzębia. Co więcej, większość badaczy zaprzecza związkowi polimorfizmu genu interleukiny
1 z uogólnionym agresywnym zapaleniem przyzębia
u rasy kaukaskiej [35, 36].
Podobną rolę do IL-1 pełni działająca przez
te same receptory interleukina 18 (IL-18). Badania przeprowadzone w ośrodku szczecińskim potwierdziły brak związku polimorfizmu genów kodujących interleukinę 18 w agresywnym zapaleniu
przyzębia [37]. Wykazano, że częstość alleli genotypów w grupie z agresywnym zapaleniem przyzębia oraz u osób z przewlekłym zapaleniem przyzębia nie różniła się w porównaniu z grupą kontrolną. Rozkład badanych hallotypów promotora genu
IL-18 u chorych z zapaleniami przyzębia i osób ze
zdrowym przyzębiem był zbliżony. Do takich samych wniosków doszli inni autorzy [38].
Badania Sanches-Hermandeza et al. [39] potwierdziły, że stężenie IL-18 w tkankach dziąsła
osób z agresywnym zapaleniem przyzębia, przewlekłym zapaleniem przyzębia i u osób zdrowych
był podobny. Stężenie IL-12 w surowicy krwi osób
z uogólnionym agresywnym zapaleniem przyzębia
(GAgP) było większe niż u osób z przewlekłym zapaleniem przyzębia (CP) i zdrowym przyzębiem.
Również w tkankach dziąsła stężenie IL-12 było
większe u osób z GAgP w porównaniu z pacjentami zdrowymi, ale podobne u osób z CP.
Interleukina 13 jest wydzielana przede wszystkim przez limfocyty CD4, ale także przez eozynofile. Wykazuje ona podobne działanie do IL-4,
która pobudza podział limfocytów B, wywołuje
przekształcanie się limfocytów B w komórki plazmatyczne oraz odgrywa ważną rolę w odpowiedzi na pasożytnicze nicienie. Interleukiny te są
związane z alergią. Polimorfizm genu kodującego IL-13 zdaje się sprzyjać powstawaniu uogólnionego agresywnego zapalenia przyzębia u Tajwańczyków. Związek ten był istotny statystycznie tylko
w grupie osób niepalących [40].
Badania Erciyasa et al. [41] sugerują, że szybka utrata tkanek przyzębia w uogólnionym agresywnym zapaleniu przyzębia nie jest związana
z polimorfizmem genów kodujących IL-6, IL-10,
IFN-gamma i TGF-ss1, lecz z polimorfizmem genu TNF-alfa.
Bardzo przydatny do dalszych rozważań
nad ryzykiem wystąpienia agresywnych i prze-
100
E. Dembowska, M. Wiernicka-Menkiszak, R. Samulak-Zielińska
wlekłych zapaleń przyzębia jest przeprowadzony przez Stabholza et al. [42] przegląd piśmiennictwa na temat wpływu czynników środowiskowych i genetycznych.
Jednym z głównych udowodnionych czynników ryzyka agresywnego zapalenia przyzębia jest
wrodzony czynnik genetyczny, co potwierdza częste rodzinne występowanie tego schorzenia. Badania przeprowadzone w rodzinach w stanie Wirginia w 1994 r. przez Marazita et al. [43] były jak
do tej pory badaniami na największej liczbie osób.
Wykazały one, że rodzeństwo młodych pacjentów
z zaawansowanymi postaciami zapaleń przyzębia także często cierpiało na ciężkie formy zapaleń przyzębia. To pozwala przypuszczać, że agresywne postacie zapaleń przyzębia są dziedziczone
w sposób autosomalny dominujący u Afroamerykanów i osób rasy kaukaskiej z penetracją około 70%. W 2010 r. Rapp et al. [44] przeprowadzili
w Brazylii badanie licznej rodziny składającej się
z 23 osób. Matka 10 dzieci straciła zęby w młodości z powodu rozchwiania, ojciec z powodu próchnicy, u 5 z 10 rodzeństwa stwierdzono objawy
uogólnionego agresywnego zapalenia przyzębia.
Co ciekawe, wszystkie przypadki choroby wystąpiły tylko u córek. Mężczyźni w tej rodzinie byli
wolni od zapalenia przyzębia. Częstość występowania tej choroby w rodzinie wyniosła 50%. Jest to
badanie jednostkowe, zważywszy jednak na rzadkie występowanie uogólnionego agresywnego zapalenia przyzębia i dużą liczbę członków rodziny
bardzo cenne.
Starania podejmowane w ciągu ostatnich 20 lat
nie dają zatem jednoznacznej odpowiedzi na temat
wpływu specyficznych kombinacji genów na powstawanie agresywnych zapaleń przyzębia. Dodatkowo, badania nad przewlekłymi zapaleniami (CP) przyzębia także nie wykluczają wpływu
czynników genetycznych na powstawanie i rozwój
tej periodontopatii. Michalowicz et al. [45] stwierdzili bowiem, że w przewlekłym zapaleniu przyzębia może istnieć także wysoka agregacja rodzinna
38–82%. A zatem, o ile rodzinna agregacja schorzenia może być kryterium diagnostycznym, o tyle w świetle nowych badań nie jest pewnym kryterium różnicującym GAgP z przewlekłym zapaleniem przyzębia.
Badania przeprowadzone na 10 parach bliźniąt monozygotycznych i 8 dizygotycznych wskazują na brak zgodności w odniesieniu do zaawansowania przewlekłych form zapaleń przyzębia
wśród par bliźniąt [46]. Są to jednak badania przeprowadzone na zbyt małej liczbie bliźniąt. Badania przeprowadzone na 74 rodzinach (475 osób)
przez de Carvalho et al. [47] potwierdziły wpływ
czynnika genetycznego na powstawanie uogólnionego agresywnego zapalenia przyzębia. Sugerują,
że uogólnione agresywne zapalenie przyzębia jest
wywołane nie przez jeden gen, lecz przez kilka genów o stosunkowo niewielkiej ekspresji. Mogą być
one uaktywniane pod wpływem czynników środowiskowych, a innym razem bez ich udziału.
Zła higiena jamy ustnej, palenie tytoniu, czynniki
psychologiczne, status społeczny w takim samym
stopniu mogą pogarszać stan przyzębia w zapaleniach przyzębia przewlekłych i agresywnych [42].
Podsumowanie
Kliniczne rozróżnienie uogólnionego agresywnego zapalenia przyzębia i zaawansowanego uogólnionego przewlekłego zapalenia przyzębia jest trudne. Istnieją bowiem pacjenci u których agresywne i przewlekłe zapalenia przyzębia
(według definicji z 1999 r.) mają podobne objawy i przebieg. Podobnie skład mikrobiologiczny
głębokich kieszonek może budzić wątpliwości co
do różnic jakościowych [14–27]. Potwierdzeniem
trudności diagnostycznych jest to, że niekiedy diagnoza wstępna GAgP musi zostać zweryfikowana
podczas prowadzonego leczenia.
Niezaprzeczalny jest fakt, że uogólnione agresywne zapalenie przyzębia jest skutkiem nieprawidłowości w odpowiedzi immunologicznej na
czynnik infekcyjny. Prowadzi ona do utraty kontroli nad mechanizmami naprawczymi tkanek
przyzębia. Udokumentowane do dzisiaj zaburzenia odpowiedzi immunologicznej dotyczą zmniejszenia możliwości fagocytozy drobnoustrojów
przez granulocyty obojętnochłonne oraz zmniejszonego wydzielania przeciwbakteryjnego białka
hCAP-18/LL-37 przez te komórki. Nieprawidłowość ta ma najprawdopodobniej charakter wtórny, gdyż czynność neutrofilów może powracać do
normy po przeprowadzonym leczeniu [48].
Oczywiste jest także zachwianie równowagi
między cytokinami wydzielanymi przez limfocyty i monocyty/makrofagi. Skutkiem tego ostatniego jest zaburzenie niezwykle skutecznego, skomplikowanego i czułego systemu powiązań między
komórkami układu odpornościowego. W uogólnionym agresywnym zapaleniu przyzębia w porównaniu z zapaleniami przewlekłymi stwierdza
się zmniejszone wydzielanie przeciwzapalnej IL-10
i zmniejszone stężenie przeciwciał klasy IgG przeciwko A.a. i P.g. z jednocześnie zwiększoną liczbą wymienionych periopatogenów. Stężenie innych cytokin jako indykatorów GAgP jest natomiast dyskusyjne.
Wpływ genów na powstawanie zarówno przewlekłych, jak i agresywnych zapaleń przyzębia jest
pewny. Nie opisano jednak, w jaki sposób regulują
one ekspresję choroby.
Uogólnione agresywne zapalenie przyzębia
Trudności diagnostyczne nie są jedynym problemem, gdyż istnieją duże różnice w rokowaniu
co do przebiegu schorzenia oraz rezultatu leczenia. W praktyce leczenie pacjentów z uogólnionym agresywnym zapaleniem przyzębia może być
mało skuteczne. Leczenie GAgP za pomocą usuwania złogów nazębnych (skaling i root planing)
nie eliminuje takich bakterii, jak Pg i Aa z tkanek ani z krwi obwodowej [49]. Dlatego lecze-
101
nie uogólnionego agresywnego zapalenia przyzębia polega na skojarzeniu leczenia chirurgicznego
z ogólną antybiotykoterapią. Znane są jednak różnice w odpowiedzi gospodarza na zastosowane leczenie [48]. Prowadzi się dalsze badania nad rolą odpowiedzi immunologicznej gospodarza, aby
wyjaśnić przyczyny tych różnic. Możliwe, że nowe
osiągnięcia mogą doprowadzić do dalszych zmian
w klasyfikacji chorób przyzębia.
Piśmiennictwo
[1] Armitage G.C.: Periodontal diagnoses and clasification of periodontal diseases. Periodontology 2000, 2004, 34, 9–21.
[2] Demmer R.T., Papapanou P.P.: Epidemiologic patterns of chronic and aggressive periodontitis. Periodontology
2000, 2010, 53, 28–40.
[3] Levin L., Baer V., Lev R., Stabholz A., Ashkenazi M.: Aggressive periodontitis among young Izraeli army personel. J. Periodotol. 2006, 77, 1392–1396.
[4] Górska R., Pietruska M., Dembowska E., Wysokińska-Miszczuk J., Włosowicz M., Konopka T.: Częstość
występowania chorób przyzębia u osób w wieku 35–44 w populacji dużych aglomeracji miejskich. Dent. Med.
Prob. 2012, 49 (w druku).
[5] Waldrop T.C., Makler B.F., Schur P., Killoy W.: Immunologic study of human periodontitis (juvenile periodontitis). J. Periodontol. 1981, 52, 8–15.
[6] Liljenberg B., Lindhe J.: Juvenile periodontitis. Some microbiological, histopatological and clinical characteristics. J. Periodontol. 1980, 7, 48–56.
[7] Johnson R.J., Matthews J.L., Stone M.J., Hurt W.C., Newman J.T.: Immunopathology of periodontal disease.
Immunologic profiles in periodontitis and juvenile periodontitis. J. Periodontol. 1980, 51, 705–712.
[8] Fine D.H., Greene L.S.: Microscopic evaluation of root surfach assotiation in vivo. J. Periodontol Res. 1984, 19, 152–157.
[9] Macler B.F., Frosta K.B., Robertson P.B., Levy B.M.: Immunoglobulin bearing lymfocytes and plasma cells in
human periodontal disease. J. Periodont. Res. 1977, 12, 37–45.
[10] Seymour, G.J., Greenspan J.S.: The phenotypic characterization of lymphocyte subpopulations in established human periodontal disease. J. Periodontal Res. 1979, 14, 39–46.
[11] Ryder M.I.: Comparison of neutrofil function in aggressive and chronic periodontitis. Periodontology 2000, 2010,
53, 124–137
[12] Smith M., Seymour G.j., Cullinan M.P.: Histopathological features of chronic and aggressive periodontitis. Periodontology 2000, 2010, 53, 45–54.
[13] Artese L., Simon M.J., Piattelli A., Ferrari D.S., Cardoso L.A., Faveri M., Onuma T., Piccirilli M., Perrotti V., Shibili J.A.: Immunohistochemical analysis of inflammatory infiltrate in aggressive and chronic periodontitis: a comparative study. Clin. Oral Invest. 2011, 15, 233–224.
[14] Socransky S.S., Haffajee A.D.: Dental biofilms difficult therapeutic targets. Periodontology 2000, 2002, 28, 12–55.
[15] Nędzi-Góra M., Kowalski J., Krajewski J., Górska R.: Analiza mikrobiologiczna głębokich kieszonek
przyzębnych u osób z przewlekłym zapaleniem przyzębia metodą PCR. Czas. Stomatol. 2007, 30, 717–725.
[16] Rakić M., Zelić K., Pawlica D., Hadzimihajlović M., Milasin J., Milcić B., Nikoli N., Stamatović N.,
Matić S., Aleksie Z., Janković S.: Association between clinical parameters and presence of Aggregatibakter actinomycetemcomitans and Porphyromonas gingivalis in patients with progressive periodontal lesions. Vonosanit
Pregl. 2010, 11, 898–900.
[17] Casarin R.C., Ribeiro Edel P., Mariano F.S., Nociti F.H.Jr., Casati M.Z., Goncalves R.B.: Levels of Aggregatibacter actinomycetemcomitans, Porphyromonas gingivalis, inflammatory cytokines and species-specific immunoglobulin G in generalized aggressive and chronic periodontitis. Periodontal Res. 2010, 45, 635–642.
[18] Cortelli J.R., Roman-Torres C.V., Aquino D.R., Franco G.C., Costa F.O., Corelli S.C.: Occurrence of Aggregatibacter actinomycetemcomitans in Brazilian with chronic periodontitis. Braz. Oral Res. 2010, 24, 217–223.
[19] Imbronito A.V., Okuda O.S., Maria de Freitas N., Mortira Lotufo R.F., Nunes F.D.: Detection of herpesviruses and periodontal pathogens in subgingival plaque of patients with chronic periodontitis, generalized aggressive periodontitis or gingivitis. J. Periodontol. 2008, 79, 2313–2320.
[20] Matarazzo F., Ribeiro A.C., Feres M., Faveri M., Mayer M.P.: Diversity and quantitative analysis of Archaea
in aggressive periodontitis and periodontaly healthy subjects. J. Clin. Periodontol. 2011, 38, 621–627.
[21] Gajardo M, Silva N., Gόmez L., Leόn R., Parra B., Contreras A., Gamonal J.: Prevalence of periodontopathic bacteria in aggressive periodontitis patients in Chilean population. J. Periodontol. 2005, 76, 289–294.
[22] Lafaurie G.I., Contreras A. Barόn A., Botero J., Mayorga-Fayad I., Jaramillo A., Giraldo A., González
F., Mantilla S., Botero A., Archila L.H., Diaz A., Chacόn T., Castillo D.M., Betancourt M., Aya M.D.R.,
Arce R.: Demographic, clinical and microbiological aspect of chronic and aggressive periodontitis in Colombia:
a multicenter study. J. Periodontol. 2007, 78, 629–639.
[23] Riep B., Edesi-Neuss L., Claessen F., Skarabis H., Ehmke B., Flemming T.F., Bernimoulin J-P., Göbel U.B.,
Moter A.: Are putative periodontal pathogens reliable diagnostic markers? J. Clin. Microbiol. 2009, 47, 1705–1711.
102
E. Dembowska, M. Wiernicka-Menkiszak, R. Samulak-Zielińska
[24] Faveri M., Mayer M.P.A., Feres M., de Figueiredo L.C., Dewhirst F.E., Faster B.J.: Microbiological diversity
of generalized aggressive periodontitis by I6S rRNA clonal analysis. Oral Microbiol. Immunol. 2008, 23, 112–118.
[25] Armitage G.C.: Comparison of the microbiological features of chronic and aggressive periodontitis. Periodontology 2000, 2010, 53, 70–88.
[26] Schlafer S., Riep B., Griffen A.L., PetrichA., Hübner J., Friedmann A., Göbel U.B., Moter A.: Filifactor alocis-involvement in periodontal biofilms. BMC Microbiol. 2010, 1, 10–66.
[27] Urzúa B., Hermosilla G., Gamonal J., Morales-Bozo I., Canals M., Barahona S., Cóccola C., Cifuentes V.:
Yeast diversity in the oral microbiota with periodontitis Candida albicans and Candida dubliniensis colonize the
periodontal pockets. Med. Mycol. 2008, 46, 783–793.
[28] Eick S., Pfister W., Sigusch B., Straube E.: Phagocytosis of periodontopathogenic bacteria by crevicular granulocutes in depressed in progressive periodontitis. Infect. 2000, 28, 301–304.
[29] Türkoğlu O., Kandiloğlu G., Berdeli A., Emingil G., Atilla G.: Antimicrobial peptide hCAP–18/LL–37 and
mRNA expresion In different periodontal diseases. Oral Dis. 2011, 17, 60–66.
[30] Dimou N.L., Nicolopoulos G.K., Hamadrakas S.J., Bagos P.G.: Fc-gamma receptor polimorphisms and their
assotiation with periodontal disease: a meta-analysis. J. Clin. Periodontol. 2010, 37, 255–265.
[31]Chrzęszczyk D., Konopka T.: Znaczenie Toll-podobnych receptorów w patogenezie zapaleń przyzębia. Dent.
Med. Prob. 2009, 46, 94–103.
[32] Teles R.P., Gursky L.C., Faveri M., Rosa E.A., Teles F.R., Socranscy S.S., Haffajee A.D.: Realionships between subgingival microbiota and GCF biomarkers in generalized aggressive periodontitis. J. Clin. Periodontol.
2010, 37, 312–323.
[33] Rescal B., Rosalem W. jr., Teles R.P., Fischer R.G., Haffajee A.D., Socransky S.S., Gustafsson A., Fiqueredo C.M.: Immunologic and microbiologic profiles of chronic and aggressive periodntitis subjecta. J. Periodontol. 2010, 81, 1308–1316.
[34] Baradaran-Rahimi H., Radar M., Arab H.R., Tavacol Afshari J., Ebadian A.R.: Association of interleukin–1 receptor antagonist gene polymorphisms with generalized aggressive periodontitis in an Iranian population.
J. Periodontol. 2010, 81, 1342–1346.
[35] Grigoriadou M.E., Koutayas S.O., Madianos P.N., Strub J.R.: Interleukin-1 as a genetic marker for periodontitis: a review of the literature. Quintes. Int. 2010, 41, 517–525.
[36] Scapoli C., Borzani I., Guarnelli M.E., Mamolini E., Annunziata M., Guida L., Trombelli L.: IL-1 gene luster is not linked to aggressive periodontitis. J. Dent. Res 2010, 98, 457–461.
[37] Droździk A., Górnik W., Kurzawski M., Dembowska E., Trąbska-Świstelnicka M., Banach J.: Wpływ
genu interleukiny 18 (IL-18) na ryzyko wystąpienia zapalenia przyzębia. Czas. Stomatol.2008, 61, 473–479.
[38] Nares S.: The genetic relationship to periodontal disease. Periodontology 2000, 2003, 32, 36–49.
[39] Sanches-Hermandez P., Zamora-Perez A., Feuntes-Lerma M., Roblem Gomez C., Mariaud-Schmidt R.,
Guerrero-Velazquez C.: IL12 and IL18 levels in serum and gingival tissue in aggressive and chronic periodontitis. Oral Dis. 2011, 18, 10.1111/j1601-0825.2011.011798.
[40] Wu Y.M., Chuang H.L., Ho Y.P., Tai C.C.: Investigation of interleukin–13 gene polymorphismus in individuals with chronic
and generalized aggressive periodontitis in Taiwanese (Chinese) population. J. Periodontal Res. 2010, 45, 695–701.
[41] Erciyas K., Pehlivan S., Sever T., Igci M., Arslan A., Orbak R.: Association between TNF-alfa, TGF-beta1,
IL–10, IL–6 and IFN-gamma gene polymorphisms and generalized aggressive periodontitis. Clin. Invest. Med.
2010, 33, E85.
[42] Stabholz A., Sokolne W.A., Shapira L.: Genetic and enviromental risk factors for chronic periodontitis and aggressive periodontitis. Periodontology 2000, 2010, 53, 138–153.
[43] Marazita M.I., Burmeister J.A., Gunsolley J.V., Koertge T.E., Lake K., Schenkein H.A.: Evidence for autosomal
dominant inheritance and race-specific heterogenity in early onset periodontitis. J. Periodontol. 1994, 65, 623–630.
[44] Rapp G.E., Pineda-Truillo N., McQuillin A., Tonetti M.: Genetic power of a Brazilian three-generation family with generalized aggressive periodontitis. Braz. Dent. J. 2010, 21, 137–141.
[45] Michalowicz B.S., Diehl S.R., Gunsolley J.C., Sparks B.S., Brookes C.N., Koertge T.E., Califano J.V., Burmeister J.A., Schenkein H.A.: Evidence of a substantial genetic basis for risk of adult periodontitis. J. Periodontol. 2000, 71, 1699–1707.
[46] Torres de Heens G.L.: Monozygotuc twins are discordant for chronic periodontitis. J. Periodontol. 2010, 37, 120–128.
[47] De Carvalho F.M., Tinoco E.M.B., Govil M., Marazita M.L., Vieira A.R.: Aggressive periodontitis is likely
influenced by a few small effect genes. J. Clin. Periodontol. 2009, 36, 468–473.
[48] Konopka T., Ziętek M.: Współczesne poglądy na rolę zaburzeń czynnościowych granulocytów obojętnochłonnych
w etiopatogenezie zapaleń przyzębia. Dent. Med. Probl. 2002, 39, 117–126.
[49] Castillo D.M., Sánches-Beltrán M.C., Castellanos J.E., Sanz I., Mayorga-Fayad I., Sanzm., Laufarie
G.L.: Detection of specific periodontal microorganisms from bacteraemia after periodontal therapy using molecular-based diagnostics. J. Clin. Periodontol. 2011, 38, 418–427.
Adres do korespondencji:
Elżbieta Dembowska
Zakład Periodontologii PUM
ul. Powstańców Wlkp. 72
70-111 Szczecin
tel.: 91 466 17 45, faks: 91 466 17 44, e-mail: [email protected]
Praca wpłynęła do Redakcji: 8.11.2011 r.
Po recenzji: 2.12.2011 r.
Zaakceptowano do druku 9.03.2012 r.
Received: 8.11.2011
Revised: 2.12.2011
Accepted: 9.03.2012