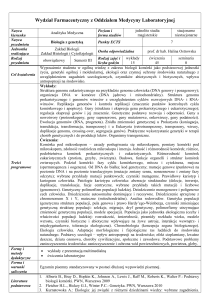
ALEKSANDRA ŚWIERCZ
Różnorodność osobników gatunku
Single Nucleotide Polymorphism (SNP)
◦ Różnica na jednej pozycji, małe delecje, insercje (INDELs)
◦ SNP pojawia się ~1/1000 pozycji
◦ Można je znaleźć porównując odczyty z jednego osobnika do genomu
referencyjnego
Structural variations to duże różnice w genomach. Mogą to być duże:
◦
◦
◦
◦
Delecje – brak fragmentu genomu
Insercje – wstawienie fragmentu genomu
Inwersje – odwrócenie fragmentu genomu
Translokacje – zmiana położenia fragmentu genomu (może być również na
innym chromosomie)
◦ Duplikacje – powtórzenia fragmentów genomu
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
2
SNP
Niedopasowania
SNP
Screen z mapowania IGV
Błąd
sekwencjonowania
SNP
homozygota
SNP
heterozygota
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
3
Który z osobników jest rodzicem,
a który dzieckiem?
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
4
R.Nielsen, JS.Paul, A.Albrechtsen, YS.Song „Genotype and SNP calling from next-generation sequencing data”
Nature Reviews Genetics 12, 443-451 (2011)
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
5
dbSNP oraz HapMap
dbSNP – obecnie 139 wersja bazy. Pojawiły się nowe organizmy wraz z
listą różnic (dotychczas zbadaną) między osobnikami
HapMap – międzynarodowy projekt, który ma na celu wykrycie i
skatalogowanie podobieństw i różnic pomiędzy organizmami ludzkimi.
Ośrodki biorące udział w projekcie pochodzą z Japonii, Wielkiej Brytanii,
Kanady, Chin, Nigerii oraz Stanów Zjednoczonych.
Projekt HapMap jest ogólnodostępny, i ma na celu pomoc środowisku
biomedycznemu w znalezieniu genów powodujących choroby i
odpowiedzi na leki terapeutyczne.
W bazie HapMap analizowano DNA z 270 osobników populacji
Afrykańskiej, Azjatyckiej i Europejskiej. Badano zarówno osobników
pojedynczych, jak i trio, czyli rodziców wraz z ich dorosłym potomkiem
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
6
Różnice strukturalne SV
Monya Baker „Structural variation: the genome's hidden architecture” Nature Methods 9,133–137 (2012)
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
7
Sposoby na wykrywanie SV
RD – Read Depth – badanie głębokości pokrycia
RP – Read Pairs – sprawdzenie mapowania odczytów sparowanych, czy
mapują się z taką samą odległością (wg. rozkładu), czy mapują się w
odpowiednią stronę, czy mapują się oba odczyty z pary
SR – Split Reads – szukanie odczytów, które nie mapują się w całości do
genomu referencyjnego, lecz jego fragmenty mapują się w odległych
miejscach – odczyty te świadczą o nietypowym (innym niż w gen. ref.)
połączeniu między fragmentami genomu
AS – AsseMbly de novo – asemblacja de novo (bez mapowania)
odczytów, następnie porównanie, czy zasemblowane kontigi pokrywają
się z genomem referencyjnym
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
8
Jakie SV można odkryć dzięki różnym
podejściom?
Monya Baker „Structural variation: the genome's hidden architecture” Nature Methods 9,133–137 (2012)
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
9
R.E. Mills et al., Mapping copy number variation by population-scale genome sequencing, Nature 470, 59–65, 2011
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
10
Read Depth
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
11
Kropka zielona to średnie pokrycie odczytami
dla okna 1kbp zdrowej tkanki pacjenta
Kropka czerwona to średnie pokrycie odczytami
dla okna 1kbp chorej tkanki pacjenta
Takie same falowanie kropek zielonych i
czerwonych oznacza, że nie ma różnic między
zdrową i chorą tkanką (tego samego) pacjenta,
tylko są różnice między genomem pacjenta, a
genomem referencyjnym
Zmiana w zachowaniu między kropkami zielonymi
i czerwonymi oznacza zmianę liczby kopii danego
fragmentu
Średnie pokrycie dla zdrowej tkanki jest ok. 52,
natomiast dla chorej tkanki ok. 40
A. Świercz
Dzięki analizie zmiany głębokości pokrycia można
znaleźć jedynie różnice w liczbie kopii
poszczególnych fragmentów, nie wiemy jednak nic
na temat położenia fragmentów w genomie.
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
12
Głębokość pokrycia – wykrywanie duplikacji
Badany genom
Genom referencyjny
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
13
Wykrywanie SV za pomocą odczytów sparowanych
Badany genom
Długość fragmentu
Genom referencyjny
Odległość mapowania
Brak różnic w strukturze genomu badanego i referencyjnego, gdyż:
◦ Długość fragmentu jest taka sama jak odległość mapowania na genomie referencyjnym
◦ Odczyty są zmapowane na genomie referencyjnym zgodnie z oryginalnym fragmentem
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
14
Insercja
insert
Badany genom
Długość fragmentu
Genom referencyjny
Odległość mapowania
Insercja w genomie badanym, gdyż:
◦ Odległość mapowania w genomie referencyjnym jest mniejsza niż długość fragmentu
długość insertu = długość fragmentu - odległość mapowania (± rozrzut długości)
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
15
Insercja – przypadek czy na pewno?
insert
Badany genom
Długość fragmentu
Genom referencyjny
Odległość mapowania
Spójność przy mapowaniu innych odczytów w tym miejscu
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
16
Insercja – przypadek czy na pewno?
insert
Badany genom
Długość fragmentów
Genom referencyjny
Odległość mapowania
Długość fragmentów nie jest równa, może się różnić ± 10% (zależy od
przygotowania biblioteki)
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
17
Insercja – przypadek czy na pewno?
insert
?
Badany genom
Długość fragmentu
Genom referencyjny
Odległość mapowania
Zbyt długi fragment wziął udział w sekwencjonowaniu, a reszta odczytów nie
potwierdza insercji.
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
18
Inwersja
Badany genom
Długość fragmentu
Genom referencyjny
Odległość mapowania = m
|m – długość fragmentu| < długość inwersji
Fragment genomu uległ inwersji, gdyż:
◦ Odczyty zmapowane są na genomie referencyjnym odwrotnie (discordant)
◦ Długość fragmentu oraz odległość mapowania są różne (to nie jest konieczne!)
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
19
Inwersja – jaka jest długość fragmentu?
Badany genom
Długość fragmentu
Genom referencyjny
Odległość mapowania = m
|m – długość fragmentu| < długość inwersji < |m+ długość fragmentu|
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
20
Inwersja – spójność mapowania
Badany genom
xa
x'a
xb
x'b
Genom referencyjny
Odległość mapowania A
Odległość mapowania B
Odległość mapowania A = odległość mapowania B
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
21
Inwersja
Badany genom
Genom referencyjny
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
22
Delecja
Badany genom
Długość fragmentu
Genom referencyjny
Odległość mapowania
Fragment genomu uległ delecji w genomie badanym, gdyż:
◦ Długość fragmentu jest krótsza niż odległość mapowania na genomie referencyjnym
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
23
Translokacja, duplikacja ?
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
24
Split reads
Odczyty, które nie mapują się w całości do genomu referencyjnego, lecz
jego fragmenty zmapowane w odległych miejscach świadczą o
rearanżacjach chromosomowych
Sekwencjonowanie =>
odczyty sparowane
Mapowanie do
genomu
referencyjnego
Odczyty
zmapowane do
genomu
A. Świercz
Odczyty
niezmapowane –
mogą leżeć na
łączeniu wariantów
strukturalnych
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
25
Split reads
Ht-seq_2012_module3.pdf
Canadian Bioinformatics Workshop
www.bioinformatics.ca
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
26
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
27
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
28
Jak duże są różnice SV?
Ile SV jest pomiędzy dwoma osobnikami?
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
29
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
30
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
31
Różnice między ludźmi
Ludzie różnią się:
kilkoma tysiącami delecji
Kilkuset duplikacjami
Kilkuset inwersjami
Kilkuset insercjami transpozonów
Kilkuset przesunięciami genów
W wynikach różnych metod do wykrywania SV mamy:
Wiele błędów pozytywnych
Wiele błędów negatywnych
Trudności w wykryciu SV, które nachodzą na sekwencje repetytywne
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
32
False positives
http://www.completegenomics.com/FAQs/CNV-Analysis/
Walidacja innymi metodami, w celu likwidacji błędów false positives
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
33
Wpływ wariantów strukturalnych na dawkę genów
Feuk, L. et al. Structural variation in the human genome. Nature Review Genetics 7, 92 (2006)
Did you know that a large number of your genes exist in variable numbers of copies? While they can
overlap with disease-related genes, these variants exist in healthy individuals too.
Większość zmian liczby kopii można znaleźć w zdrowych osobnikach. Podejrzewa się że te zmiany
powodują choroby poprzez szereg mechanizmów – pokazanych na rysunku.
Po pierwsze różna liczba kopii może
spowodować różną dawkę genu
poprzez delecje lub insercje, które
może spowodować że odmienny gen
ulegnie ekspresji – potencjalnie
powodując chorobę.
Dawka genu opisuje liczbę kopii
genu w komórce, co się przekłada na
zwiększoną lub zmniejszoną
ekspresję tego genu.
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
34
Delecje mogą spowodować zmniejszoną dawkę
genu, poprzez usunięcie jednego allelu
lub poprzez delecję allelu (dominującego)
ujawni się recesywna wersja genu.
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
35
Jeśli warianty strukturalne nakładają się na
geny, to może zostać zredukowana lub w ogóle
zablokowana ekspresja genu poprzez inwersję,
translokację czy delecję.
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
36
Warianty SV mogą także mieć wpływ na
elementy regulatorowe, jeśli zostanie on
usunięty może zostać zwiększona lub
zmniejszona ekspresja genu.
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
37
Czasami kombinacja dwóch lub większej liczby wariantów
może spowodować złożoną chorobę, podczas gdy
pojedyncze zmiany nie powodują żadnego efektu.
Dodatkowo złożone choroby mogą się pojawić jeśli różna
liczba kopii jest połączona z innymi genetycznymi lub
środowiskowymi czynnikami.
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
38
Podsumowanie
Różne podejścia do sekwencjonowania wysokoprzepustowego:
Wady i zalety metod
Długość odczytów
Jakość sekwencji na końcówkach sewkencji
Odczyty sparowane, pojedyncze
Specyficzne rodzaje błędów
Mapowanie do genomu referencyjnego:
Algorytmy dopasowania lokalnego, globalnego i semiglobalnego
Macierze kropkowe (dotmatrix)
Tworzenie indeksu BWT
Haszowanie
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
39
Podsumowanie 2
Asemblacja de novo
Trudności w asemblacji
Powtórzenia zaburzają obliczenia
Overlap–layout–consensus
Grafy de Bruijna(błędne ścieżki w grafach)
Wady i zalety obu podejść
RNA-sequencing:
Algorytmy mapowania sekwencji RNA (różne podejścia: asemblacja de novo,
mapowanie do transkryptomu, mapowanie do genomu)
Przeszkody w mapowaniu RNA do genomu
Trudność w rozpoznawaniu nowych transkryptów
Różnicowa ekspresja genów i alternatywny splicing(warianty splicingowe)
A. Świercz
ANALIZA DANYCH WYSOKOPRZEPUSTOWYCH
40