Choroby metaboliczne
wrodzone
Rok akademicki 2008/2009 K.Pawelec
Choroby metaboliczne
wrodzone
Wszystkie choroby genetyczne w których
uchwytne odchylenia biochemiczne są
wynikiem mutacji monogenowej.
Mutacja prowadzi do zmian w produkcji
białka (enzymu, nośnika) którego brak lub
zmiana aktywności prowadzi do
upośledzenia danej reakcji czy procesu
metabolicznego
Objawy kliniczne występujące w
genetycznych chorobach
metabolicznych
Uszkodzenie czynności wątroby:
żółtaczka, zburzenia krzepnięcia
Hepatomegalia i hepatosplenomegalia
Wymioty
Zaćma, zwichnięcie soczewki
Zmętnienie rogówki
Zwyrodnienie barwnikowe siatkówki
„ Wiśniowa „ plamka na dnie oka
Dysmorfia twarzy
Objawy kliniczne występujące w
genetycznych chorobach
metabolicznych
Anomalie układu kostnego
Zespół Fanconiego-De Toniego-Debrego
Kardiomiopatia
Kamica nerkowa
Niedokrwistość
Zespół Rey’a
Wyprysk skórny
Zespoły wad wrodzonych
Epizody intoksykacji
Objawy kliniczne występujące w
genetycznych chorobach
metabolicznych
Opóźnienie rozwoju umysłowego
Drgawki
Wiotkość
Makrocefalia
Postępujące zmiany demielinizacyjne
Postępujące uszkodzenie OUN
Neuropatia obwodowa
Objawy biochemiczne stwierdzane
w genetycznych chorobach
metabolicznych
Hipoglikemia
Kwasica metaboliczna
Hiperamonemia
Acydemia mleczanowa
Hiperurikemia
Objawy biochemiczne stwierdzane
w genetycznych chorobach
metabolicznych
Zaburzenia krzepnięcia i
hiperbilirubinemia
Wzrost kinazy kreatyniny
Substancje redukujące w moczu
Ketonuria
Neutropenia
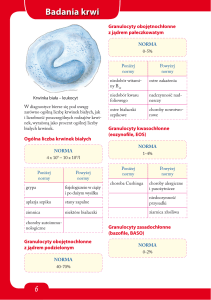
Badania wykonywane w celu
rozpoznania chorób metabolicznych
Pełna morfologia
Mocznik
Elektrolity
Luka anionowa
Gazometria krwi
Glukoza
Mleczany
Wskaźniki biochemiczne czynności wątroby
substancje redukujące w moczu
Ciała ketonowe w moczu
Mleczany w płynie m-r
Nieprawidłowy zapach moczu i ciała w
chorobach metabolicznych
Mysi
fenyloketonuria
(kwas fenylooctowy)
Syropu klonowego
choroba syropu
klonowego(MSUD)
Kapusty, zjełczałego masła
tyrozynemia typ I
Spoconych stóp
acydemia izowalerianowa
Kwaśny
acydemia metylomalonowa
Kociego moczu
3-metylokrotonyloglicynuria
Siarkowy
cystynuria
Rybi
trimetyloaminuria,
dimetyloglicynuria
Nieprawidłowy kolor moczu
w chorobach metabolicznych
Czarnobrązowy
alkaptonuria
Czerwonobrązowy
mioglobinuria, hemoglobinuria
Czerwony
porfirie
Pomarańczowy
zespół Lescha-Nyhana
(moczany)
Diagnostyka wrodzonych chorób
metabolicznych
Nadmiar metabolitu będącego substratem zablokowanej
reakcji biochemicznej (fenyloalanina w fenyloketonurii)
Patologiczne metabolity w wyniku reakcji alternatywnych
(kwas fenylopropionowy w fenyloketonurii)
Brak produktu zablokowanej reakcji (tyrozyny w
fenyloketonurii)
Oznaczenie aktywności enzymatycznej lub wykazanie
nieobecności białka
Wykazanie mutacji genowej
Diagnostyka wrodzonych chorób
metabolicznych
Testy przesiewowe
Phenistix (fenyloketonuria) substancje redukujące w
moczu
Test Meyera (cystynuria i homocystynuria)
Chromatografie AA metodą jakościową i półilościową w
surowicy o moczu (aminoacydemie i aminoacydurie)
Chromatografia monocukrów w moczu (fruktozemia,
galaktozemia)
Oligosacharydy w moczu (choroba Pompego)
Mukopolisacharydy (mukopolisacharydozy)
Diagnostyka wrodzonych chorób
metabolicznych
Testy wysokospecjalistyczne
Ilościowe oznaczanie AA w surowicy i moczu metodą
chromatografii (fenyloketonuria) - substancje redukujące w
moczu
Badanie profilu kwasów organicznych w moczu metodą
chromatografii gazowej ( kwasice organiczne)
Aktywność enzymów w erytrocytach (galaktozemia)
Aktywność enzymów w leukocytach ( choroby
liposomalne)
Diagnostyka wrodzonych chorób
metabolicznych
Testy wysokospecjalistyczne
Aktywność enzymów w limfocytach ( zaburzenia oksydacji kwasów tłuszczowych)
Aktywność enzymów w tkankach ( fenyloketonuria)
Testy obciążeniowe
Badanie mutacji genowej
Badanie rodzinne w kierunku heterozygoty
Leczenie i zapobieganie
wrodzonym chorobom
metabolicznym
Eliminacja substratu z pokarmów poprzez stosowanie
odpowiedniej diety
Substytucyjne podanie produktu reakcji
Próby uzupełnienia brakującego enzymu
Przeszczep
Testy skriningowe (nie mogą dawać wyników fałszywie
ujemnych) w Polsce fenyloketonuria i niedoczynność
tarczycy w okresie noworodkowym ( 5 doba)
Poradnictwo genetyczne
Choroby metaboliczne
Zaburzenia przemiany węglowodanów
Zaburzenia metabolizmu aminokwasów
Acydemie organiczne
Hiperamonemie wrodzone
Zaburzenia beta-oksydacji kwasów
tłuszczowych
Choroby metaboliczne
Hiperlipidemie
Choroby mitochondrialne
Choroby liposomalne
Choroby peroksymalne
Zaburzenia przemiany
węglowodanów
Galakotozemia klasyczna
Dziedziczna nietolerancja fruktozy
Glikogenozy
Galaktoza
Galaktozemia klasyczna
Mutacja genu urydylilotransferazy galaktozo-1fosforanu ( 70% mutacja zmienia sens odczytu
Q188R)
Po karmieniu mlekiem ( matki lub krowim) wymioty
biegunka, hepatosplenomegalia, żółtaczka,
kwasica, zaburzenia krzepnięcia, hipoglikemia,
cukromocz, pogorszenie stanu ogólnego,
uszkodzenie OUN i kanalika proksymalnego,
zaćma.
Galaktozemia klasyczna
Badanie przesiewowe przy urodzeniu
w suchej kropli – ilościowe oznaczenie aktywności
enzymu: urydylilotransferazy galaktozo-1fosforanu ( w erytrocytach, fibroblastach) gdy
transfuzja krwi badanie dopiero po 3 miesiącach
na diecie eliminacyjnej
Podwyższony poziom galaktozy w moczu i krwi
(gdy podawana laktoza), hipoglikemia, kwasica
mleczanowa, podwyższony poziom transaminaz,
zaburzenia krzepnięcia, dodatni test w moczu na
substancje redukujące.
Galaktozemia klasyczna
Dieta – wyłączenie laktozy na całe życie
Preparaty mlekozastępcze : sojowe, hydrolizaty
kazeiny
Po diecie znaczna poprawa stanu ogólnego
i ustąpienie objawów
Mimo diety odległe następstwa: niedobory
wzrostu, zaburzenia rozwoju intelektualnego
i mowy, niewydolność hormonalna jajników
Fruktoza
Dziedziczna nietolerancja fruktozy (HFIhereditary fructose intolerance) mutacja genu
aldolazy B- fruktozo-1-fosforanu, gen
zlokalizowany na chromosomie 9q13-32,
najczęstsze mutacje (A149P-67% alleli i
AQ174D 16% alleli)
Objawy podobne do galaktozemii, pojawiają się
dopiero po podaniu soków, owoców, sacharozy i
miodu, rzadko przed 3 mż
Dziedziczna nietolerancja fruktozy
(HFI)
Wymioty, zahamowanie rozwoju
psychoruchowego i fizycznego, uszkodzenie
nerek i wątroby
Hipoglikemia, kwasica, ostre lub przewlekłe
objawy intoksykacji, uszkodzenie kanalika
proksymalnego z objawami zespołu
Fanconiego-De Toniego-Debrrego, w moczu
substancje redukujące ( fruktoza) często
rozpoznawane zakażenie z bakteriemią
Dziedziczna nietolerancja fruktozy
(HFI)
Oznaczenie aktywności enzymu w wątrobie lub
ostrożne wykonanie testu obciążenia fruktozą
(objawowa hipoglikemia i kwasica
mleczanowa)
Dieta eliminacyjna bez fruktozy. U pacjentów
występuje awersja do słodkich pokarmów,
owoców , jarzyn.
Pacjenci z HFI nie mają próchnicy zębów.
Glikogen
Glikogenoza typu I
Glikogenozy typu III i VI (IX)
Inne glikogenozy
Glikogenoza typu I
Deficyt glukozo-6-fosfatazy (GSC Ia - glicogen
storage disease, von Gierke)
Glikogenoza typu Ib- brak swoistej traslokazy i
glukoza-6-fosforan nie przedostaje się do
mikrosomów markerem tego typu jest
neutropenia z ropniami przewodu pokarmowego
i martwiejące zapalenie jelita.
Patomechamizm spowodowany hipoglikemią
(niski poziom insuliny, zwiększenie poziomu
glukagonu, kortyzolu, insuliny, hormonu wzrostu)
Glikogenoza typu I
Objawy: bóle brzucha ( powiększenie wątroby i
nerek), zahamowanie wzrostu, przyrost masy
ciała, policzki „jak u lalki”, hipoglikemia po 1-2
godz od posiłku, kwasica metaboliczna,
hiperlipoproteoinemia, hiperurikemia, ketonuria
Rozpoznanie: glukagon podany dożylnie
(0,03mg/kg) brak wzrostu glukozy -płaska
krzywa cukrowa, obciążenie glukozą - krzywa
cukrzycowa, badanie aktywności glukozo-6fosfatazy w bioptacie wątroby typ Ia, translokazy
w typie Ib ( świeża tkanka)
Glikogenoza typu I
Leczenie: utrzymanie normoglikemii (60120mg%) - częste karmienie, roztwór glukozy iv,
starsze dzieci -dodawanie do posiłków skrobi
kukurydzianej
Przeszczep wątroby
Rokowanie 1/3 niemowląt i małych dzieci ginie z
powodu hipoglikemii i zakażeń ( typ Ib)
Zaburzenia metabolizmu
aminokwasów
Fenyloketonuria
Tyrozynemia typu I
Albinizm
Klasyczna homocystynuria
Choroba syropu klonowego
Fenyloalanina
Fenyloketonuria ((PKU)brak aktywności
hydrolazy fenyloalaniny ( brak przejścia w
tyrozynę)
Pierwsze objawy w 3 mż - zahamowany
rozwój psychoruchowy, wymioty, wysypki
skórne, jaśniejsza karnacja skóry, drgawki
nadpobudliwość, hipotonia mięśniowa,
małogłowie „mysi zapach„
Fenyloalanina
Podwyższony poziom fenyloalaniny w
surowicy ( <20mg%) i w moczu łącznie z
kwasem fenylopropionowym (z chlorkiem
żelaza - zielony kolor moczu )
Gen zlokalizowany na chromosomie 12
częstość występowania 1:7500-8000
Test przesiewowy Guthriego(do 1997) - inhibicja
bakterii ( fenyloalanina pobudza bakterie do
wzrostu) od 1997 test ilościowy kolorymetryczny
firmy IBL wszystkie noworodki krew na bibułę
Fenyloalanina
Potwierdzenie rozpoznania po (+) teście
przesiewowym w specjalnych ośrodkach do 2
mż
Leczenie : dieta eliminacyjna najpóźniej od 3mż,
mieszanki mlekozastępcze bez zmniejszenia
podaży białka(Nofelan, Nofemix, Lofenale)
Poziom fenyloalaniny w czasie diety 3-7mg% w
surowicy , dieta przez całe życie.
Gdy za mało AA: zahamowanie wzrostu,
niedokrwistości biegunki, zgon.
Fenyloalanina
Matka z fenyloketurią w ciąży poziom AA <10mg%,
częste poronienia
Fenyloketonuria matczyna : opóźnienie umysłowe,
małogłowie, i/lub wrodzona wada serca
Inne postaci fenyloketonurii: złośliwa
hiperfenyloalaninemia - deficyt tetrahyrobiopteryny
(BH4)- objawy nie odróżnienia od PKU ale nie
ustępują po klasycznej diecie ( oznaczenie
neopteryny i biopteryny w moczu, test obciążeniowy
BH4, )
Rokowanie ostrożne mimo wczesnego rozpoznania i
wprowadzenia leczenia substytucyjnego
neurotrasmiteramii
Fenyloalanina
Hiperfenylolaninemia łagodna - częściowe
obniżenie aktywności hydroksylazay
fenyloalaniny
Podwyższenie poziomu fenyloalaniny <20mg%
Zwykle nie wymaga ścisłej diety
Może prowadzić do fenyloketonurii matczynej
Hiperfenyloalaninemia przejściowa - okresowe
zwiększenie stężenia fenyloalaniny w surowicy i
moczu noworodka- opóźnienie dojrzałości
enzymatycznej
Tyrozyna
Tyrozynemia typu I
Albinizm
Tyrozynemia
Tyrozynemia typu I
deficyt hydrolazy fumaroacetioctanu, gromadzi
się bursztynyloaceton- bezpośrednio uszkadza
narządy ( wątroba, nerki)
Ostra tyrozynemia pierwsze m-ce życia, szybki
zgon, uszkodzenie wątroby( żółtaczka
wodobrzusze, zaburzenia krzepnięcia,
hipoglikemia), uszkodzenie kanalika
nerkowego, (białkomocz, hiperfosfaturia,
glikozuria, aminoacyduria- z. F-De T-D)
Tyrozynemia typu I
Postać przewlekła ciężka krzywica witamio-D zależna,
przewlekła neuropatia obwodowa, ( obraz podobny do
porfirii)
W 30% przypadków rozrost nowotworowy w wątrobie
carcinoma hepatocellulare
Rozpoznanie podwyższony poziom w surowicy i
moczu tyrozyny, w moczu kwas parahydroksyfenylopirogronowy (PHPP i jego pochodne)
Podstawa rozpoznania wykrycie bursztynylooctanu w
surowicy i moczu - GC-MS- (gas chromatographymass spectrometry)
Tyrozynemia typu I
Leczenie podawanie inhibitora oksydazy PHPP preparat NTBC , który zapobiega gromadzeniu się
bursztynyloacetonu w wątrobie i nerkach, preparat
nasila hipertyrozynemię i musi być stosowany z
ograniczeniem spożycia tyrozyny i fenyloalaniny
W czasie leczenia może dochodzić do powikłań
skórno-ocznych (odkładanie kryształków tyrozyny w
tkankach)
monitorowanie poziomu AA w surowicy ,, okresowo
alfa-fetoproteinę w surowicy (co 3-4m-ce) USG
brzucha , TK, ewentualnie przeszczep wątroby
Metionina
Klasyczna homocystynuria - brak
aktywności enzymu syntetyzującego
cystationinę z homocysteiny i seryny.
Upośledzenie dojrzewania kolagenuzburzenia układu kostnego z osteoporozą,
zwichnięcie soczewek, zakrzepy w
naczyniach, w niektórych postaciach
choroby skuteczne jest leczenie vit B6
Klasyczna homocystynuria
Pacjenci mają jasną karnację, delikatną skórę, są
szczupli, spłaszczenie kręgów, koślawość kolan,
zniekształcenie klatki piersiowej, chód kaczkowaty,
rozwój umysłowy niekiedy upośledzony
Rozpoznanie: test Meyera z cyjanonitroprusydkiemczerwone zabarwienie na obecność aminkwasów
siarkowych, zwiększone stężenie homocysteiny i
metioniny, a zmniejszone cystyny w surowicy i moczu
Ostateczne rozpoznanie deficyt aktywności syntetazy
cystationiny w wątrobie lub fibroblastach skóry.
Należy ocenić witaminozależność podejmując próbę
leczenia Vit B6
Leucyna, izoleucyna, walina
Choroba syropu klonowego
(MSUD- maple syrup urine disease)
Zaburzenia dekarboksylacji ketokwasów
pochodzących z dezaminacji leucyny,
izoleucyny i waliny powoduje to ciężki
zespół u noworodka - intoksykacji - zgon
lub ciężkie upośledzenie rozwoju
psychoruchowego
Choroba syropu klonowego
W klasycznej postaci MSUD noworodek zdrowy
w 1 tyg ż pogorszenie stanu og (wiotkość,
drżenia, senność, śpiączka), hipoglikemia,
ketony w moczu, kwasica metaboliczna, zgon
we wczesnym niemowlęctwie
Postać przerywana MSDU w różnym wieku :
utrata przytomności, drgawki, ataksja, infekcje w
stanach zwiększonego katabolizmu
Wariant MSDU zależny od wit B1 -zmniejszona
aktywność dekarboksylazy rozgałęzionych
ketokwasów do wiązania konenzymu tej reakcji
Choroba syropu klonowego
Rozpoznanie - podwyższony poziom leucyny ,
waliny i izoleucyny i zwiększone wydalanie AA w
moczu metodą GC-MS i testy enzymatyczne w
w leukocytach, mocz - zapach karmelu
zawsze sprawdzić w MSDU czy objawy ustępują
po vit B1
Dieta eliminacyjna i zbilansowana
W pierwszym okresie dializy ,
Gdy leczenie późno uszkodzenie OUN
Choroby lizosomalne
U podstaw tych chorób leży w odkładaniu
nadmiaru wielkocząsteczkowych
związków w lizosomach z powodu braku
aktywności jednego z enzymów,
dziedziczenie autosomalne recesywne
Postępujące uszkodzenie komórek
poprzez powiększone lizosomy,
spichrzanie obejmuje : układ nerwowy,
kostny, narządy miąższowe, rogówkę
tkankę podskórną,
Choroby lizosomalne
W zależności od rodzaju spichrzanej
substancji wyróżnia się:
Mukopolisacharydozy,
Sfingolipidozy,
Gangliozydozy,
Sulfatydozy,
Sialidozy,
Spichrzanie glikoprotein
Choroby lizosomalne
Lipidoza - choroba Gauchera
Deficyt beta-glukozydazy glukozyloceramidu,
postać bez objawów neurologicznych -typ 1
dorosłych przebieg przewlekły,
hepatosplenomegalia, hipersplenizm,
niedokrwistość, żółtaczka, osteoporoza)
postać neuropatyczna typ 2- niemowlęcy (objawy w
pierwszych miesiącach życia, szybki zgon,
narastające uszkodzenie OUN, wyniszczenie)
postać neuropatyczna typ 3-młodzieńczy powolny
przebieg
Choroby lizosomalne
Lipidoza - choroba Gauchera
Leczenie sztucznie zmodyfikowany enzym
ludzkiej beta glukozydazy (ceradaza)
skuteczny w postaciach bez objawów
neurologicznych , nie przenika do płynu m-r
Pierwsza choroba lizosomalna z leczeniem
przyczynowym.
Hiperlipidemie
Zwiększone stężenie lipoprotein w
surowicy- przedwczesna miażdżyca
naczyń, zawał, zatory, udary mózgu,
dziedzicznei monogenowe, dominujące
Przykłady
Rodzinna hipercholesterolemia
Deficyt lipazy lipoproteinowej
Rodzinna hipercholesterolemia
Upośledzenie wiązania receptorowego
lipoprotein LDL
Częstość występowania heterozygot 1:500
Objawy kliniczne w dzieciństwie tylko u homozyt
(1:1mln)- żółtaki skóry, ścięgien, choroba
wieńcowa, nadciśnienie, zgon przed 20 rż
Heterozygoty nie mają objawów klinicznych w
dzieciństwie rozpoznanie na podstawie
>220mg% cholesterol u dziecko i jednego z
rodziców, u homozyt >600mg%
Ostateczne rozpoznanie -zmniejszone
Rodzinna hipercholesterolemia
Ostateczne rozpoznanie -zmniejszone
powinowactwo receptorów LDL w hodowli
fibroblastów skóry lub badanie DNA w kierunku
mutacji genu receptora,
Diagnostyka prenatalna - płyn owodniowy
Leczenie utrzymanie wartości cholesterolu poniżej
220mg% ( ograniczenie spożywania tłuszczów-1520g/dobę, cholestyramina 0,6g/kg/dobę), u
homozygot - inhibitory reduktazy kwasu
hydroksymetyloglutarowego (HMGA) -wastatyna,
kwas nikotynowy, selektywna plazmafereza,
przeszczep wątroby
Choroby peroksymalne
U podstaw tych chorób leży nieprawidłowa
biosynteza komórkowych organelii
zawierajacych katalazę tzw:
peroksysomów lub brak poszczególnych
enzymów tych organelli : enzymów
peroksymalnych ( uczestniczą w beta oksydacji kwasów tłuszczowych o bardzo
długich łańcuchach -VLCFA)
Zespół Zellwegera
Zespół mózgowo-wątrobowo-nerkowy
Ujawnia się przy urodzeniu
Wysokie czoło, duże ciemię, niedorozwój łuków
brwiowych, zmarszczka nakątna, hipotonia
mięśni, drgawki , upośledzony rozwój
psychoruchowy, hepatomegalia zaburzenie
gospodarki żelazem, zwyrodnienie barwnikowe
siatkówki- zgon w 1 rż
Zwiększone stężenie kwasów
tłuszczowych o długim łańcuchu(VCFA)
brak peroksysomów ( biopsja wątroby)
Choroba Lescha-Nyhana
Deficyt fosforybozylotransferazy
hipoksantynoguaninowej (HGPRT) enzymu
uczestniczącego w metaboliźmie puryn
cecha recesywna sprzężona z chromosomem X
u chłopców w 3-4mż opóźnienie rozwoju psychruchowego, mocz pomarańczowy, kamica
nerkowa, ubytki tkanek
Podwyższony poziom kwasu moczowego ,
aktywność HGPRT w krwinkach czerwonych
Choroba Wilsona
Zwyrodnienie wątrobowo- soczewkowe
Dziedziczenie autosomalne recesywne
Upośledzone wiązanie miedzi w surowicy, obniżenie
poziomu ceruloplazminy (poniżej 20mg%), odkładanie
miedzi w tkankach i zwiększone wydalanie.
Postępujące uszkodzenie wątroby, drżenia, dysartia, ruchy
choreoatetotyczne (po 10 rż)
Pierścień Kaysera-Feischera
Penicylamina – usuwa nadmiar miedzi i zapobiega
gromadzeniu,
Sole miedzi doustne w celu zmniejszenia wchłaniania
jelitowego
Mukowiscydoza
Torbielowate zwłóknienie trzustki (cystis
fibrosis)
Dziedziczenie autosomalne recesywne
Gen mukowiscydozy kodujący białko CFTR
(budowa kanału chlorowego)- ramię długie
chromosomu 7
Patogeneza- lepkość śluzu, zaburzenie
transportu chloru
Mukowiscydoza
Zmiany niedodmowo- rozedmowe i zapalne płuc,
Zmiany torbielowate trzustki z upośledzeniem
wydzielania enzymów trawiennych i
przewlekłymi zaburzeniami wchłaniania
Zespół zagęszczonej żółci i zwłóknienie wątroby,
Zwiększona zawartość chlorków w pocie,
Postać: płuca, brzuszna i mieszana
Mukowiscydoza
Test potowy – zwiększona zawartość chlorków
pocie,
Identyfikacja mutacji genu CFTR- biologia
molekularna,
Korelacja genotypu z postacią kliniczną
Antybiotykoterapia, fizykoterapia, uzupełnianie
enzymów trzustkowych, płyny, amilorid
(transport sodu)
Lokalna terapia genowa