Choroby metaboliczne
Anna Materna-Kiryluk
Katedra i Zakład Genetyki Medycznej UM w Poznaniu
Chorobę metaboliczną uwzględnić należy:
•U wszystkich noworodków z niejasną, poważną lub postępująca chorobą szczególnie, gdy
przebieg ciąży lub porodu był prawidłowy
•U wszystkich dzieci z ostrym pogorszeniem stanu ogólnego i lub ograniczeniem świadomości
szczególnie, gdy poprzedzone były wymiotami, gorączką lub głodzeniem
•U wszystkich dzieci z objawami kwasicy i hipoglikemii
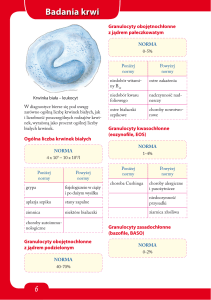
Podstawowe badania metaboliczne w stanach nagłych:
•Pobranie krwi do oznaczeń (cito): elektrolity, glukoza, CRP, CK, ALAT, ASPAT,
kreatynina, mocznik, kwas moczowy, gazometria, wskaźniki krzepnięcia
•Amoniak, mleczan
•Zachowanie próbki osocza na oznaczenie aminokwasów, acylokarnityn
•Zachować bibułę z kroplą wysuszonej krwi na oznaczenie acylokarnityn, aminokwasów
(TANDEM MS), ew. analizy DNA
Podstawowe badania metaboliczne c.d
•Badanie moczu (ciała ketonowe, glukoza , białko, pH
•(uwaga pH>5 w czasie kwasicy (kanalikowa kwasica nerkowa)
•Pobrać próbkę moczu z ostrej fazy na kwasy organiczne i dodatkowe testy metaboliczne
(GCMS)
•Zachować płyn mózgowo-rdzeniowy (natychmiast zamrozić)
Skrining selektywny w kierunku chorób metabolicznych
przy podejrzeniu zaburzeń metabolicznych
-Metoda MS/MS
(analiza suchej kropli krwi na bibule metodą tandemowej spektrometrii mas)
-Metoda GC-MS
(Analiza profilu kwasów organicznych w moczu metodą GC-MS)
Postępowanie w metabolicznych stanach nagłych
• Rozpocząć wlew 10% roztworu glukozy 150ml/kg/d z właściwą podażą elektrolitów
•Kontrolować poziom mleczanów i gazometrii
•Skontaktować się z Ośrodkiem Chorób Metabolicznych (Centrum Zdrowia Dziecka- Warszawa)
Fenyloketonuria
•Testy przesiewowe na fenyloketonurię opierają się na analizie poziomu stężenia fenyloalaniny
we krwi (metoda kolorymetryczna).
•Krew powinna być pobrana rano, na czczo. Pobiera się ją z palca ręki, z pięty lub z płatka ucha.
•Choroba genetyczna, uwarunkowana autosomalnie recesywnie
•Spowodowana mutacjami w genie hydroksylazay fenyloalaniny (PAH)
•Efektem mutacji jest podwyższone stężenie fenyloalaniny we krwi i jej metabolitów w moczu
chorych
•Częstość występowania od 1:10 000 do 1:20 000 żywych urodzeń
Fenyloketonuria – objawy:
•stopniowo rozwijające się opóźnienie rozwoju umysłowego
•wymioty
•jasna skóra i niebieskie oczy
•stęchły mysi zapach
•drgawki (25%)
•zmiany w EEG (50%)
•inne: małogłowie, wydatna szczęka górna, szerokie rozstawienie zębów, hipoplazja szkliwa i
opóźnienie wzrostu
Fenyloketonuria – objawy u dzieci chorych matek
•opóźnienie rozwoju fizycznego i psychoruchowego
•wady wrodzone
•małogłowie
niska urodzeniowa masa ciała
Galaktozemia
•Wrodzony błąd metaboliczny uniemożliwiający przekształcenie galaktozy w glukozę.
Gromadzący się na skutek istnienia bloku metabolicznego galaktozo-1-fosforan wywiera
działanie toksyczne.
Galaktozemia
•Choroba genetyczna, uwarunkowana autosomalnie recesywnie
•Mutacje w genie GALT
•W moczu stwierdza się obecność cukrów (galaktozy),
•We krwi brak aktywności enzymu urydilotransferazy galaktozo-1-fosforanu
•Częstość występowania w Europie 1:55 000 żywo urodzonych dzieci
Galaktozemia - objawy
•uszkodzenie wątroby
•uszkodzenie nerek
•uszkodzenie soczewki oka
•uszkodzenie układu nerwowego
Galaktozemia - leczenie
• Polega na szybkim wprowadzeniu diety bezgalaktozowej (bezmlecznej) – preparaty bez
galaktozy i laktozy
• Powikłaniami w przypadku galaktozemii mogą być opóźniony rozwój psychiczny i dysfunkcja
gonad.
Podejrzewając zaburzenia przemiany galaktozy należy najpierw zaprzestać karmienia
mlekiem, a następnie diagnozować dziecko!!!
Choroby lizosomalne
W okresie noworodkowym objawy mało wyraźne:
•Początkowo hipotonia mięśniowa, opóźnienie motoryki i upośledzenie umysłowe
•Postępująca utrata nabytych umiejętności
•Postępująca organomegalia
•Pogrubiałe rysy twarzy, zmiany w kośćcu deformacje kostne), zmiany skórne
•Ataksja, nadpobudliwość, spastyczność
•Wiśniowa plamka na dnie oka w niektórych chorobach
Zespół Smitha-Lemlego-Opitza
•Zaburzenie endogennej syntezy cholesterolu
•Mutacja w genie(DHCR7)
•Brak lub zmniejszona aktywność reduktazy 7 dehydrocholesterolu(DHCR7)
•Zmniejszenie stężenia cholesterolu
•Kumulacja bezpośrednich prekursorów cholesterolu (7- i 8- DHC)
•Zaburzenie przebiegu embriogenezy
Objawy zespołu Smitha-Lemlego-Opitza:
Cechy dysmorfii twarzy: krótki zadarty nos, małożuchwie
Małogłowie lub wady strukturalne mózgowia
Rozszczep podniebienia
Wada serca, niedorozwój/brak nerek
Nieprawidłowości zewn. narządów płciowych
Polidaktylia pozaosiowa dłoni i/lub stóp, syndaktylia 2
i 3 palca stóp
Obniżone napięcie mięśni
Słaby przyrost masy ciała