Podstawowe informacje o chemii
Rozpoczynając szkolną naukę chemii dobrze wiedzieć o paru sprawach, które w szkolnej nauce nie
są podawane w ogóle, albo mówi się o nich zdecydowanie za późno i za mało.
Pod pojęciem chemii rozumiemy naukę o procesach przebiegających z wytworzeniem lub
zerwaniem wiązania chemicznego. Najczęściej mamy do czynienia z procesami, w których
następują oba te zjawiska. Przemiany, w których nie ma zmian w obrębie wiązań, należą do fizyki.
Skoro o chemii decydują wiązania, należałoby zacząć naukę od ich opisania. Ale tu „zaczynają się
schody”. Aby choć trochę pojąć istotę wiązań chemicznych, należy poznać budowę i właściwości
atomów. Natomiast żeby poznać i wyjaśnić właściwości chemiczne atomów i ich skłonność do tworzenia wiązań, trzeba posłużyć się mechaniką kwantową, która do prostych i łatwych niestety nie
należy. Dochodzi więc do paradoksalnej sytuacji, że aby ułatwić poznanie i zrozumienie chemii,
trzeba by zacząć od arcytrudnej mechaniki kwantowej oraz budowy materii (też niezbyt łatwej, o
czym za chwilę). I to zapętlenie jest prawdopodobnie powodem, że w dydaktyce chemii od stulecia
nic nie drgnęło. Początki nauki są dokładnie takie same jak w ubiegłym wieku, mimo że o podstawach chemii wiemy o niebo więcej niż wówczas, gdy te zręby chemicznej dydaktyki powstawały.
Żeby przeciąć ten gordyjski węzeł, proponuję zapoznanie się z naprawdę niezbędnym minimum
wiedzy dotyczącej świata atomów. Ze względu na trudności w wyjaśnianiu tego świata mikro za
pomocą prostych i jasnych sformułowań, musisz na początek wiele twierdzeń przyjąć na wiarę.
Czas na rzetelne wyjaśnienie problemów związanych z mechanika kwantową przyjdzie ewentualnie
później, po przebyciu całego kursu chemii. A skoro nie będziemy w sposób pełny wyjaśniać praw
świata atomów, to przynajmniej należy Ci się wyjaśnienie, czemu jest to takie trudne.
Od urodzenia poznajemy otaczający nas świat za pomocą zmysłów, a więc poznajemy zjawiska,
procesy, które są skutkami, a nie przyczynami. Później zaczyna się wyjaśnianie przyczyn tych znanych nam już skutków, ale umysł wyćwiczony w poznawanie skutków, często właśnie przez ich
pryzmat próbuje widzieć przyczyny (a nie odwrotnie). To, że Ziemia przyciąga nas i wszystkie
przedmioty grawitacją przyjmujemy za prawdę dość łatwo (bo takie są nasze doświadczenia) ale to,
że my również przyciągamy Ziemię z taką samą siłą, już tak łatwo nas nie przekonuje. Na temat
tego, co się dzieje w świecie cząsteczek i atomów bezpośrednio naszymi zmysłami nie jesteśmy w
stanie dowiedzieć się nic. Wszystko (a jest tego sporo) co o tym świecie wiemy, pochodzi z obserwacji pośrednich, skutków makroskopowych i wyciąganych na tej podstawie wniosków. A sprawa
jest jeszcze bardziej pogmatwana, bo nie dość, że nie widzieliśmy ani nie dotknęliśmy nigdy pojedynczej cząsteczki czy atomu (o elektronach nie wspominając) to jeszcze wnioski, które wyciągamy z obserwacji pośrednich, są tak zaskakujące i nieprzystające do naszego obrazu makroświata, że
„zwykłego” człowieka przyprawiają o ból głowy. No ale w końcu inni sobie z tym poradzili (przynajmniej niektórzy) - spróbujmy i my!
Budowa atomu:
Jądra atomów, z których zbudowana jest cząsteczka związku chemicznego, składają się z dodatnio naładowanych protonów i obojętnych elektrycznie neutronów. Siły elektrostatycznego odpychania jednoimiennych ładunków powinny powodować nietrwałość takiego tworu - jądro powinno „eksplodować” a odpychające się dodatnie protony z dużą prędkością powinny oddalać się od siebie. Ponieważ tak się nie dzieje, wnioskujemy stąd, że w jądrze atomowym działają
jeszcze jakieś siły utrzymujące jego spójność, mimo owych destrukcyjnych sił odpychania. Są to tzw. siły bliskiego zasięgu, działające na bardzo krótkich odległościach. Jeżeli przyjmiemy, że średnica protonu czy neutronu wynosi około
-15
1 fermi (10
m) to zasięg działania sił krótkiego zasięgu, spajających jądro, wynosi około 2-3 fermi. Tak więc siły te
działają wyłącznie na najbliższe sobie nukleony i praktycznie zasięg ich działania ogranicza się do jądra. Odpychanie
elektrostatyczne protonów jądra działa na odległości znacznie większe (maleje z kwadratem odległości między protonami) lecz jest o wiele słabsze, najczęściej pomijalnie małe w porównaniu z siłami spójności. Jednak przy odległościach
między protonami rzędu 0,5 fermi (protony sąsiadujące ze sobą) wielkość sił odpychania elektrostatycznego zaczyna
równoważyć siły przyciągania nukleonów. Mamy tu do czynienia z przykładem stabilnej równowagi między dwiema
przeciwstawnymi siłami (stan w przyrodzie bardzo często spotykany).
Tak więc w jądrze atomowym „zmagazynowana” jest olbrzymia ilość energii (olbrzymia w porównaniu do masy jądra).
Możemy porównać jądro atomowe do butli z gazem pod dużym ciśnieniem, gdzie zmagazynowaną energię możemy
wyzwalać powoli, pod kontrolą, spalając gaz w kuchence (reaktor atomowy, kontrolowany rozpad jąder) lub może nastąpić niekontrolowane wyzwolenie energii (wybuch butli), co w odniesieniu do jąder atomowych oznacza wybuch jądrowy (bomba atomowa). Poza dość wąską dziedziną chemii jądrowej, chemików ten zakres opisu materii interesuje
mniej niż inne - jest to raczej dziedzina pracy fizyków.
Orbitale atomowe:
Chemik jądro atomowe najczęściej traktuje jako monolityczny twór o określonych właściwościach (nie wolno jednak
zapominać, że te właściwości to wypadkowa właściwości i zachowań składowych) i bardziej niż „wnętrze” interesują
go relacje jądra z pozostałymi składowymi atomu, czyli z elektronami. Tu znów znajdujemy równowagę między siłami
elektrostatycznego przyciągania (dodatnie jądro - ujemne elektrony) i elektrostatycznego odpychania (elektrony między
sobą). Im elektrony są bliżej jądra, tym mniejsze odległości między nimi i silniejsze odpychanie - zarówno przez elektrony powłok bliższych jądra, jak i elektronów danej powłoki. Przy pewnej odległości następuje zrównoważenie sił
przyciągania przez dodatnie jądro i odpychania przez pozostałe elektrony atomu.
Pojawia się tu jednak problem, trudny do wyjaśnienia na gruncie elektrostatycznych oddziaływań - dlaczego elektrony
powłoki najbliższej jądra (1s), które są spychane przez inne elektrony w kierunku jądra, i które przez to jądro są przyciągane, utrzymują się jednak w pewnej, dość ściśle określonej odległości od niego? I tu koniecznym staje się zastosowanie zasad i praw mechaniki kwantowej, która wykaże, że tak musi być, ale nie wyjaśni nam niestety w prosty sposób,
dlaczego tak musi być.
Zdajemy już sobie sprawę, jak skomplikowany układ sił działa na elektron w atomie. Siły te (elektryczne, magnetyczne)
sumując się wektorowo, powodują powstanie wokół jądra atomowego obszarów, w obrębie których na umieszczony
tam elektron oddziałują bardzo małe siły. Taki obszar, gdzie elektron poddawany jest działaniu bardzo małych sił, nazywamy orbitalem. Ponieważ w przestrzeni wokół jądra, nie należącej do orbitali, na elektron działają znaczne siły,
elektrony są „zaganiane” do obszarów orbitali. Jeżeli jednak w obrębie orbitala znajdą się choćby dwa elektrony, zaczynają oddziaływać na siebie odpychająco, a więc „wypychają się” nawzajem z tej pozycji. Ponieważ jednak elektrony
prócz pól elektrycznych oddziałują na siebie także polami magnetycznymi, powstającymi jako konsekwencja ich ruchu
wirowego (spin), dwa elektrony na jednym orbitalu niwelują odpychanie elektryczne przyciąganiem magnetycznym
(przeciwne spiny to przeciwny układ biegunów magnetycznych). Stąd jeden orbital może pomieścić najwyżej dwa elektrony o przeciwnych spinach. Wyobraźmy sobie tacę, której np. jedna czwarta powierzchni to łagodne zagłębienia. Nasypmy na tę tacę garść grochu i potrząsając delikatnie, obserwujmy, gdzie będą lokalizować się ziarna grochu. Nie jest
dla nas żadnym zaskoczeniem, że za każdym razem zdecydowana większość grochu ulokuje się w zagłębieniach. Te zagłębienia to model orbitali.
Często orbitale w podręcznikach są definiowane jako miejsca, gdzie prawdopodobieństwo znalezienia elektronu jest
największe. Czasem nawet precyzują, że owo największe oznacza 90% lub 95%. Kształt orbitala określa (fizycznie nie
istniejąca !!) granica zamykająca ów obszar 95% prawdopodobieństwa znalezienia elektronu.
Podręczniki, pokazując graficzny obraz orbitali, mówiąc o ich kształcie, wyrabiają w nas podświadome przekonanie o
fizycznym, wręcz materialnym charakterze orbitala. Jeszcze bardziej utwierdzają nas w tym podświadomym przekonaniu, mówiąc o „naprężeniach” wiązań, jeśli orbitale tworzące wiązanie chemiczne nakładają się pod innymi kątami niż
ich „naturalne” ułożenie w przestrzeni. Są to bardzo wygodne, ale bardzo nieprawdziwe modele. Co prawda często
przekonująco wyjaśniające pojedyncze zjawisko (np. nietrwałość cyklopropanu) ale jednocześnie wprowadzające zamieszanie w zrozumieniu ogólnych pojęć (np. wiązania chemicznego).
Tak więc nie wchodząc zbyt głęboko w skomplikowaną materię wszelkich sił działających w obrębie atomu i wzajemnych relacji między nimi wystarczyć nam musi, że skomplikowana gra sił grawitacji (masa), elektrycznych (ładunek) i
magnetycznych (spin) powoduje, że jądro atomowe jest tworem bardzo trwałym (do pewnej masy - atomy promieniotwórcze) a w pewnych obszarach wokół jądra atomowego siły te dla konkretnego elektronu praktycznie się znoszą. Jeżeli elektron przez przypadek znalazł się poza tym obszarem, działa zawsze na niego jakaś siła wypadkowa, kierująca
go w konsekwencji właśnie do tego obszaru o najniższej energii. Obszary te (orbitale) układają się w pewnych konkretnie określonych odległościach od jądra (powłoki elektronowe opisane główną liczbą kwantową), a w obrębie tych powłok tworzą obszary o różnym kształcie i energii (orbitale opisane poboczną liczbą kwantową - patrz rysunek niżej)
oraz ukierunkowaniu w przestrzeni (opisane magnetyczna liczbą kwantową). Tak więc „adres” konkretnego elektronu
w atomie musi składać się z podania numeru powłoki (główna liczba kwantowa, w starszych podręcznikach stosowano
też oznaczenia literowe K, L, M, N), typu orbitalu (poboczna liczba kwantowa l - poszczególnym jej wartościom przypisujemy oznaczenia literowe typów orbitali: s, p, d, f... - patrz rysunek niżej) i jego usytuowania w przestrzeni (np. p x,
py, pz) - magnetyczna liczba kwantowa. Warto tu wspomnieć, że istnienie orbitala wynika z budowy danego atomu, tak
więc dany orbital istnieje, bez względu na to czy są elektrony mogące go wypełnić (orbital obsadzony) czy też nie
(orbital nieobsadzony).
Wiązania:
Jeśli już przeanalizowaliśmy budowę atomu i pojęcie orbitali, możemy zacząć rozpatrywać problem wiązań chemicznych. Tu mamy jeszcze bardziej skomplikowany układ sił przyciągania i odpychania elektrostatycznego, bierze w nim
udział bowiem kilka atomów (a dwa co najmniej). Wiązania chemiczne na ogół są tworzone przez orbitale należące do
ostatniej, walencyjnej powłoki elektronów. Jeżeli dwa orbitale dwóch atomów („czerwony” i „niebieski” na rysunkach
poniżej) zbliżą się do siebie (rysunek pierwszy), to elektron „niebieski” znajdzie się w zasięgu działania silnych sił
„czerwonego” i odwrotnie (odpychanie elektronów). Jeśli jednak przezwyciężymy te siły i jeszcze bardziej zbliżymy
atomy do siebie, tak by przestrzeń orbitalu „niebieskiego” i przestrzeń orbitalu „czerwonego” były w dużej części
tożsame, to oba elektrony znajdą się w sytuacji minimalnych oddziaływań (mechanika kwantowa) - wspólna przestrzeń
to teraz orbital cząsteczkowy, wiążący. Tak w dużym skrócie wygląda tworzenie najpopularniejszego wiązania typu
sigma, powstające przez współosiowe nałożenie się orbitali.
Teraz w rozważania o wiązaniach wtrąćmy informacje o bardzo ważnej cesze atomów - elektroujemności ...
Elektroujemność:
Elektroujemność jest jednym z istotniejszych parametrów opisujących właściwości pierwiastków. Elektroujemność najłatwiej wytłumaczyć na prostym układzie dwóch atomów połączonych wiązaniem chemicznym. Wspólna para elektronów tworzących wiązanie może znajdować się w środku między dwoma atomami, należąc jednocześnie do obydwu
atomów (wiązanie atomowe, patrz rysunek powyżej) lub w całości należeć do jednego tylko atomu, który przyjmuje
wówczas ładunek ujemny i z drugim atomem (który oczywiście przyjmuje wówczas ładunek dodatni) łączy go wyłącznie siła przyciągania elektrostatycznego (wiązanie jonowe).
Prócz tych dwóch skrajnych położeń wspólnej pary elektronowej, w rzeczywistości występuje całe spektrum wiązań, w
których wspólna para elektronów zajmuje pozycję pośrednią między wiązaniem atomowym a jonowym. To przesunięcie elektronów wiążących jest wynikiem różnicy w sile przyciągania elektronów przez poszczególne atomy, a siłę tę
nazywamy elektroujemnością. Elektroujemność jest to zatem pewna tendencja atomu pierwiastka do przyciągania
(„zawłaszczania”) elektronów walencyjnych innych atomów. Na wielkość elektroujemności główny wpływ mają dwa
czynniki: odległość powłoki walencyjnej od jądra atomowego oraz dążenie każdego atomu do stworzenia sytuacji, w
której ostatnia obsadzona elektronami powłoka będzie zawierać osiem elektronów. Zatem wyższą elektroujemnością
będą charakteryzować się pierwiastki początkowych okresów (mały promień atomowy, silniejsze przyciąganie przez
dodatnie jądro) i końcowych grup (duża ilość elektronów walencyjnych ułatwia osiągnięcie oktetu). Zgodnie z tą regułą
najbardziej elektroujemnym pierwiastkiem jest fluor (drugi okres, siódma grupa).
Siłę elektroujemności pierwiastka określamy poprzez wartość półempirycznego parametru. Wartości tego współczynnika dla poszczególnych pierwiastków pierwszy obliczył (i przede wszystkim zaproponował sposób ich wyliczania) Pauling. Później, stosując nieco inne podejście i sposób liczenia wartości parametru elektroujemności podali Rochow i Allred. Istnieje również inna metoda liczenia zastosowana przez Mullikena. W każdej z tych metod otrzymuje się nieco
różne wartości dla tego samego pierwiastka, z czego wynika prosty wniosek, że nie należy do wartości parametru elektroujemności przywiązywać zbyt dużej wagi i traktować go należy jako parametr wyłącznie pólilościowy. Różnice pomiędzy elektroujemnością obliczoną różnymi metodami zazwyczaj nie przekraczają wartości 0,3, choć w pojedynczych przypadkach różnica ta może nawet przekroczyć wartość 1. Zakres wartości współczynnika elektroujemności zawiera się z reguły między wartościami 1 (metale) a 4 (fluor).
Elektroujemność zależy w głównej mierze od promienia atomu oraz dążenia do uzyskania trwałej konfiguracji oktetu
elektronowego (osiem elektronów na ostatniej powłoce).
Pierwszy czynnik związany jest z siłą oddziaływania dodatnio naładowanego jądra atomowego, która głównie zależy od
wielkości ładunku jądra i promienia atomu - praktycznie można przyjąć, że siła ta słabnie wraz ze wzrostem promienia
atomu.
Drugi czynnik - chęć uzyskania oktetu elektronowego, należy rozważać w układzie dwóch atomów połączonych
wiązaniem. Tu decydującą rolę odgrywa prawdopodobieństwo. Dla przykładu - w połączonych w cząsteczkę MgO
atomach większe jest prawdopodobieństwo uzyskania oktetu przez atom tlenu, który zawierając już sześć elektronów,
potrzebując przyłączenia tylko dwóch, niż przez atom magnezu, który w tym celu musiałby przyłączyć aż sześć
elektronów (ma tylko dwa walencyjne). Magnezowi łatwiej (istnieje większe prawdopodobieństwo takiego zachowania)
uzyskać oktet przez oddanie dwóch elektronów z walencyjnej powłoki M (pozostanie mu oktet na niżej połączonej
powłoce L). Stąd czasem mówimy o pierwiastkach elektrododatnich, dla podkreślenia, że łatwiej przechodzą w postać z
oktetem elektronowym przez oddanie elektronów i uzyskanie ładunku dodatniego niż przez przyłączenie elektronów
(taka „odwrotna” elektroujemność).
Elektroujemność pierwiastków (a dokładniej różnice elektroujemności między pierwiastkami połączonymi wiązaniami
chemicznymi) wpływając na wielkość polaryzacji poszczególnych wiązań, powoduje powstawanie, w elektrycznie
obojętnej cząsteczce, miejscowych ładunków dodatnich i ujemnych, co z kolei ma znaczący wpływ na reaktywność
związków chemicznych (wytwarzają się swoiste „punkty zaczepienia” dla drugiej cząsteczki substratu). Ładunki te
determinują także w znacznym stopniu właściwości fizyczne związku (np. rozpuszczalność czy temperaturę topnienia
lub wrzenia). Są też podstawowym czynnikiem określającym charakter wiązania między dwoma atomami
(kowalencyjne, jonowe ...).
Znajomość elektroujemności i jej wpływu na reaktywność i właściwości związków chemicznych jest podstawową
wiedzą, konieczną do zrozumienia chemicznych przemian i fizykochemicznych właściwości związków.
... i wróćmy do sprawy wiązań:
W bardzo dużym uproszczeniu można powiedzieć, że cząsteczka to atomy i wiązania. O ile atomy nie ulegają jakimś
zasadniczym zmianom, o tyle wiązania łączące atomy w cząsteczkę są bardzo różne i mogą ulegać zmianom pod
wpływem innych elementów cząsteczki. Wiązanie chemiczne jest realizowane przez elektrony ostatniej powłoki
elektronowej danego pierwiastka, która nosi nazwę powłoki walencyjnej. Każdy atom, poprzez tworzenie wiązań stara
się uzyskać konfiguracje oktetu (osiem elektronów) na swojej powłoce walencyjnej. „Klasyczne” wiązanie chemiczne
polega na takim zbliżeniu dwóch atomów, że odpowiednie ich orbitale powłok walencyjnych nakładają się na siebie
(część przestrzeni między atomami jest wspólna dla obydwu orbitali tworzących wiązanie) a znajdujące się na
orbitalach elektrony (po jednym na każdym orbitalu) tworzą parę o przeciwnych spinach i znajdują się w przestrzeni
wspólnej dla obu atomów. Takie wiązanie nazywamy wiązaniem atomowym, kowalencyjnym. W praktyce spotykamy
je dość rzadko - takie wiązanie występuje między atomami tego samego pierwiastka, a więc najczęściej w przypadku
pierwiastków gazowych N2, O2, Cl2 lub Br2 czy I2.
Drugim typem wiązań, najczęściej chyba spotykanym, (szczególnie w chemii organicznej) jest wiązanie atomowe
spolaryzowane. Różni się ono od opisanego powyżej jedynie tym, że ponieważ w wiązaniu biorą udział dwa atomy
różnych pierwiastków, o różnych elektroujemnościach, „środek ciężkości” ładunku elektronów wiązania jest
przesunięty w kierunku atomu o większej elektroujemności, przez co zatraca się równowaga elektryczna i w obrębie
wiązania powstaje układ biegunowy (dipol). Jeden z atomów przyjmuje ładunek δ- a drugi δ+ i choć cząsteczka jako
całość nadal jest elektrycznie obojętna, to przesunięcia ładunku w jej obrębie powodują powstanie miejsc bardziej
podatnych na reakcje chemiczne. Ponieważ powstałe ładunki nie są równe elementarnemu ładunkowi, zaznaczamy je
symbolem δ .
W dużym uproszczeniu można powiedzieć, że aktywność chemiczna cząsteczki, jej podatność na przemiany chemiczne
zależy głównie od wielkości polaryzacji wiązań, choć nie wolno nam zapominać, że znaczna rolę odgrywa tu także
przestrzenne usytuowanie poszczególnych atomów.
Jeżeli różnica wartości elektroujemności dwóch atomów tworzących wiązanie jest większa niż 1,4 dochodzi do
powstania wiązania jonowego. Wiązanie jonowe tym się różni od atomowego spolaryzowanego, że siłą wiążącą dwa
atomy nie jest już wspólnota pary elektronowej, lecz siły przyciągania elektrostatycznego dodatniego ładunku kationu
(który oddał swój elektron, lub dokładniej, któremu elektron został zabrany) i ujemnego ładunku anionu (który z kolei
powstał przez zabranie całego elektronu z kationu). Tak więc wiązanie jonowe sensu stricto nie jest wiązaniem
chemicznym, a jedynie czystym, elektrostatycznym oddziaływaniem między jonami. Takie „czyste” wiązanie jonowe
występuje równie rzadko co czyste wiązanie kowalencyjne. Zdecydowana większość wiązań chemicznych to mniej lub
bardziej spolaryzowane wiązania kowalencyjne.
Ze względu na to, że w wiązaniu jonowym następuje przeniesienie elektronu (przesunięcie całego ładunku
elementarnego), kationy i aniony zaznaczamy znakiem plus lub minus, już bez symbolu delty. Wiązania typu sigma
silnie spolaryzowane i jonowe dość łatwo doprowadzić do dysocjacji. Wiązania jonowe wymagają tylko niewielkiego
wkładu energii potrzebnego do odsunięcia jonów od siebie (pokonania jedynie sił elektrostatycznego przyciągania
odmiennych ładunków). Zupełnie inna jakość wiązania jonowego powoduje, że reakcje między związkami o
charakterze jonowym przebiegają wyraźnie inaczej niż reakcje między związkami o wiązaniach atomowych.
Dysocjacja
wiązania
„prawie”
jonowego
Wiązanie „czysto” jonowe
Ponieważ wiązania atomowe spolaryzowane możemy uznać za stan pośredni, między wiązaniem atomowym (brak
polaryzacji wiązania) a wiązaniem jonowym („pełna”, maksymalna polaryzacja) czasem w różnych opracowaniach
możemy spotkać się z określaniem procentowego udziału wiązania jonowego w wiązaniu atomowym, dla bardziej
precyzyjnego określenia charakteru wiązania spolaryzowanego. Z wiązaniem jonowym mamy do czynienia przede
wszystkim w chemii nieorganicznej.
Specyficznym rodzajem wiązania chemicznego jest wiązanie koordynacyjne, inaczej donorowo-akceptorowe. Ta
druga nazwa dokładnie oddaje charakter wiązania. Jest to wiązanie pozornie takie jak atomowa - wspólna para elektronów łączy dwa atomy - lecz różni się tym, że oba elektrony pochodzą od jednego atomu (donora) i są wykorzystywane także przez drugi atom (akceptor). Tego typu wiązania spotykamy w związkach, gdzie występują atomy na stopniu
utlenienia większym od +4.
Ponieważ generalnie elektronów walencyjnych wokół każdego atomu może być tylko osiem (w przypadku wodoru
dwa), to np. dla kwasu siarkowego(VI), trzymając się formalnych wartościowości, uzyskamy wzór (a). Wokół atomu
siarki znajduje się wówczas aż 12 elektronów walencyjnych (sześć wiązań po 2 elektrony). Jest to niezgodne z podanym wcześniej warunkiem, więc musimy przyjąć, że dwa wiązania siarki z atomami tlenu są wiązaniami donorowo
(siarka daje) akceptorowymi (tlen przyjmuje), co oznaczamy strzałkami od donora do akceptora. W takim układzie wzór (b) - wokół wszystkich atomów jest odpowiednia ilość elektronów walencyjnych (po osiem, przy wodorach po
dwa). Związki o takiej strukturze wiązań są najczęściej silnymi utleniaczami (kwas azotowy(V), nadmanganiany, kwas
chlorowy(VII) itp.)
Istnieje jeszcze jeden typ wiązań, nieco podobny do wyżej omówionego - wiązania powstające przez nałożenie się
orbitala zawierającego parę elektronów (tzw. wolną parą elektronów) i orbitala nieobsadzonego („pustego”) drugiego
atomu (p lub d). Tego typu wiązania donorowo-akceptorowe występują np. w kompleksach.
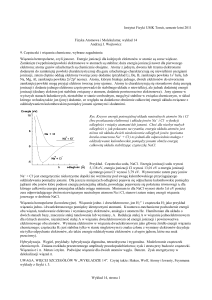
Ostatnim typem wiązania jest tzw. wiązanie wodorowe. Jest to nazwa nieco myląca, wiązanie wodorowe bowiem nie
jest wiązaniem stricte chemicznym, nie wiąże atomów w cząsteczce, jest raczej pewnym typem oddziaływania między
atomem wodoru w cząsteczce a elektroujemnym atomem innego pierwiastka, lecz ze względu na charakter tego
oddziaływania otrzymało ono nazwę wiązania. Jest to dość słabe oddziaływanie występujące pomiędzy atomem wodoru
połączonego z atomem silnie elektroujemnego pierwiastka (np. tlenu lub azotu) a elektroujemnym atomem,
posiadającym tzw. wolne pary elektronowe, tzn. elektrony z powłoki walencyjnej, niebiorące udziału w wiązaniu. „Siłą
napędową” powstania takiego wiązania jest częściowy ładunek dodatni na atomie wodoru, który powstaje wskutek
polaryzacji wiązania wodór - atom elektroujemny (najczęściej tlen, np. w grupie –O–H) i związany z tym cząstkowy
ładunek ujemny na atomie elektroujemnego pierwiastka. Jednak charakter sił wiążący atom wodoru z atomem
elektroujemnego pierwiastka nie ma charakteru oddziaływań elektrostatycznych, tak jak ma to miejsce między
dipolami, lecz posiada dość wyraźnie zaznaczony charakter wiązania chemicznego (wspólnota elektronów), aczkolwiek
niezmiernie słabego. Udział w nim biorą wolne pary elektronowe atomu silnie elektroujemnego pierwiastka. Mimo
niewielkiej siły wiązanie wodorowe jest przyczyna wielu ważnych zjawisk fizykochemicznych. Jemu zawdzięczamy
istnienie naszego życia, gdyby bowiem nie wiązania wodorowe, woda (podstawa życia na Ziemi) nie byłaby cieczą o
znanych nam właściwościach, lecz podobnie jak metan byłaby gazem, na co wskazuje jej formalna masa cząsteczkowa
(metan 16, woda 18, amoniak 17, azot 28, tlen 32). W rzeczywistości wiązania wodorowe między cząsteczkami wody
powodują, że zachowuje się ona jak substancja o masie o wiele większej (100-150). Silnie elektroujemny atom tlenu
powoduje polaryzację wiązań H–O, umożliwiając powstanie oddziaływań o charakterze wiązań wodorowych, tak jak
podano na rysunku poniżej.
Dotychczas mówiliśmy o wiązaniach typu sigma, gdy orbitale wiążące nasuwają się na siebie współosiowo. Istnieją
jeszcze wiązania typu pi, gdzie orbitale wiążące nakładają się bokami:
Tego typu wiązania pojawiają się jako drugie lub trzecie wiązanie w tzw. Wiązaniach wielokrotnych
- podwójnych (np. CO2; O=C=O) lub potrójnych. Znajomość ich właściwości i powstawania będzie
miała duże znaczenie w trakcie poznawania chemii organicznej. Np. wiązania typu π (boczne nałożenie
orbitali p) łatwiej niż σ ulegają rozerwaniu, bowiem część wspólna orbitali składowych jest mniejsza, średnia energia
elektronów wiążących jest zatem większa i niewiele energii potrzeba, by rozsunąć elektrony (każdy na "swój" orbital
(rysunek B).
http://www.mlyniec.gda.pl/~chemia/ogolna/atom.htm
(budowa i elektroujemność)
http://www.mlyniec.gda.pl/~chemia/ogolna/substancje/substancja.htm
(wstęp)
I na koniec jeszcze parę ogólniejszych uwag, które mogą przyczynić się do lepszego pojmowania i
łatwiejszego uczenia się chemii (i nie tylko).
Każda przemiana - tak reakcja chemiczna, jak i zjawisko fizyczne ("fizykochemiczne") - odbywa się z pewnym efektem
energetycznym. Jedne wiązania czy oddziaływania muszą zostać pokonane, rozerwane (potrzeba energii) by mogły
powstać nowe (odzysk energii). Zatem, czy i jak przebiegnie zjawisko (reakcja), decydować będą etapy uwalniające
lub pobierające energię. Żeby jakiś etap wymagający energii mógł zaistnieć, układ musi mieć dostęp do "zapasu"
energii. Ten zapas mógł być stworzony w poprzednim etapie, wyzwalającym energię, lub takiego zapasu nie ma
i proces się zatrzymuje. Tak więc o reakcji nie decyduje reguła, np. tlenki niemetali z wodą dają kwas, lecz siła wiązań
i oddziaływań. Dla CO2 ta reguła jest prawdziwa, dla SiO2 - już nie (siły wiązań tworzących krystaliczną strukturę
krzemionki - kwarcu na to nie pozwolą w „zwykłych” warunkach). Tak więc wiedza chemiczna to nie znajomość
regułki tylko zrozumienie istoty zachowania się materii.
Następne przyzwyczajenie, od którego dobrze jest się wyzwolić, to traktowanie symboli związków i samych związków
jako bytów tożsamych. Bardzo często zapominamy, że H2O to tylko symboliczny zapis cząsteczki wody, a nie woda
jako taka. Informacje płynące z takiego zapisu nic nam nie mówią o rzeczywistości i na przykład niezrozumiałym jest,
dlaczego cząsteczki o masie molowej 18 tworzą ciecz a nie gaz, tak jak inne substancje o podobnej masie i strukturze NH3 CH4 czy H2S. Bardzo mało w czasie nauczania chemii mówi się o wpływie formy, postaci, w jakiej występują
reagenty na reaktywność substancji. Wyjątkiem, zazwyczaj jedynym, jest węgiel, na przykładzie którego nieco szerzej
omawia się wpływ struktury na reaktywność i właściwości chemiczne (grafit i diament). Omawia się jednak wówczas
te różnice z punktu widzenia alotropii pierwiastków i mało kto uogólnia i przenosi te wiadomości na inne pierwiastki i
związki chemiczne. Nie można, z praktycznego punktu widzenia, każdej reakcji opisywać pełną, wyczerpująca
informacją, bo zagmatwalibyśmy tak jej zapis, że stałby się całkowicie nieczytelny - ale trzeba ciągle pamiętać o
symboliczności zapisu równania reakcji.
I jeszcze parę uwag, w zasadzie kierowanych do studentów - ale przeczytać chyba nie zawadzi:
Im lepiej i głębiej poznajemy świat cząsteczek, atomów i cząstek elementarnych, tym bardziej staje się on dla nas ...
niezrozumiały. Rzecz w tym, że prawa i zasady obowiązujące w tym mikroświecie (mechanika kwantowa) nie przystają w żaden sposób do naszego świata w wymiarze makro. Inaczej mówiąc, nie mamy doświadczeń życiowych, które
pozwoliłyby zjawiska mechaniki kwantowej opisać przykładami makroświata o mechanice newtonowskiej. A ponieważ elementów mikroświata nie możemy obserwować bezpośrednio, za pomocą zmysłów, nie możemy również zrozumieć jak on działa. Dla doraźnych celów dydaktycznych budujemy uproszczone modele w skali makro, które tłumacząc
jedno - zaciemniają drugie. Jeżeli w wyobraźni ucznia zaszczepimy pojęcie elektronu, jako maleńkiej kulki obdarzonej
ładunkiem, trudno będzie mu przyjąć później do wiadomości, że elektron jest falą i w dodatku może być w tym samym
czasie w dwóch różnych miejscach. Jeśli jednak nie podsuniemy mu żadnego rozwiązania, żadnego modelu z zakresu
znanego mu świata makro, w ogóle nie pojmie istoty elektronu i nie zrozumie najprostszych (jak na mikroświat) pojęć
i zjawisk, jak choćby tworzenie wiązania chemicznego. Dotarłszy jednak do pewnego poziomu wiedzy - np. kończąc
naukę a zaczynając studiowanie - musimy odważnie powiedzieć sobie DOŚĆ. Dość oszukiwania się, że elektron to
kuleczka a wiązanie chemiczne to sprężynka, dość oszukiwania siebie samego, że rozumiemy co to jest pole
grawitacyjne czy elektryczne, że pojmujemy zjawisko zamiany energii na masę i że w ogóle rozumiemy co to jest masa
i energia. Przyjąwszy sokratesowskie "wiem, że nic nie wiem" (z całym szacunkiem dla proporcji), zastanówmy się, czy
przyznawszy się do niezrozumienia i niezdolności wyobrażenia sobie, dość przecież podstawowych zjawisk i pojęć,
musimy przyjąć niemożność poznania przez nas tego świata? Odwołajmy się tu na moment do matematyki, która
bazuje na aksjomatach (np. bezwymiarowość punktu czy jeden wymiar prostej, złożonej z bezwymiarowych punktów kto to ROZUMIE !?) i pojęciach też nie dających się wyjaśnić (nieskończoność, zero, wartość nieskończenie mała ...)
uchodzi (i słusznie) za królową nauk, i bez której dochodzenie do prawdy obiektywnej jest niemożliwe w żadnej
dziedzinie wiedzy, . Tak więc przyjmując coś "na wiarę" nie obniżamy rangi naukowego poznania, jeżeli tylko cała
konstrukcja zbudowana na tych aksjomatach jest spójna i skuteczna. Jeśli przyjmiemy "na wiarę" postulat fal materii de
Broglie'a, to dzięki temu będziemy mogli przewidzieć skutecznie przebieg wielu zjawisk, niewytłumaczalnych bez
przyjęcia tej teorii. Zauważmy, że bez względu na czyją korzyść rozstrzygnie się odwieczny spór materialistów z
idealistami (czy pierwsza była materia czy idea, lub trywializując: co było pierwsze - jajko czy kura) prawa fizyki nie
zmienią się na jotę. Innymi słowy, obojętnie czy przyjmiemy prymat jajka czy kury, opis jednego i drugiego pozostanie
identyczny.
Te parę zdań powyżej to tylko wywołanie tematu, z którym każdy musi się sam uporać. Każdy sam musi przemyśleć
sprawę by zrozumieć głębie stwierdzenia "wiem, że nic nie wiem" i zrozumieć, że taka "niewiedza" to wiedza bardzo
głęboka i ułatwiająca pojmowanie nawet bardzo skomplikowanych i trudnych do przeniesienia w świat makro-modeli
zjawisk atomowo-cząsteczkowych.
Osobiście proponuję następujące podejście do sprawy:
Nie odrzucajmy porównań i przykładów spotykanych w podręcznikach i na wykładach, są to bowiem najczęściej dość
dobrze przemyślane i zazwyczaj "najlepsze z możliwych" makro-schematy mikro-zjawisk. Pamiętajmy jednak, że dotyczą one tylko tego zjawiska (lub często nawet tylko jego elementu) i nie należy ich przenosić na inne zjawiska. Elektron
jako naładowana kulka może być dobrym przybliżeniem w tłumaczeniu podstaw istoty wiązania chemicznego czy dysocjacji, nie nada się jednak zupełnie do wyjaśniania zjawiska rezonansu magnetycznego jąder (NMR). Tam bardziej
przekonującym będzie porównywanie elektronów do mgły czy chmury o różnej gęstości w różnych miejscach cząsteczki - taki model z kolei nie bardzo się przyda przy objaśnianiu wiązań. Zatem nie - elektron jest kulką, czy - elektron jest
chmurą, czy - elektron jest falą, lecz - w zjawisku X możemy sobie przyjąć, że elektron jest kulką, ale w zjawisku Y
musimy o tym "zapomnieć" i przyjąć, że jest chmurą itp. Zaś czym jest elektron w rzeczywistości ? "Wiem, że nic nie
wiem" - ale reakcje chemiczne potrafię przeprowadzić zgodnie z założeniami i procesy fizykochemiczne wykorzystuję
bez kłopotów w praktyce - a więc jednak coś wiem?...
Zapominając o złożoności realnej sytuacji reakcji nie zrozumiemy także, dlaczego raz ona zachodzi a innym razem nie,
chociaż w zapisie symbolicznym wygląda to tak samo. Na przykład niełatwo pojąć, dlaczego ketozy są reduktorami,
choć ani grupa alkoholowa ani ketonowa takich właściwości nie wykazuje i dlaczego glukoza jest reduktorem, skoro
występując w postaci cyklicznej też nie ma grupy aldehydowej. Wszelkie, nawet bardzo daleko idące uproszczenia w
tłumaczeniu i zapisywaniu chemii są konieczne i zazwyczaj przydatne - inaczej zapewne już na samym początku nauczania ucieklibyśmy z krzykiem przerażenia - nie wolno jednak ani na chwilę zapominać, że są to uproszczenia, że
rzeczywistość jest bardziej złożona. Na nauczycielach wszelkich szczebli nauczania spoczywa obowiązek wyjaśniania,
w odpowiednim momencie, co jest uproszczeniem, o czym w danej chwili nie mówimy i co sprawia, że omawiany "wyjątek" jest wyjątkiem (czyli, jak już wiemy, spełnieniem innej reguły, ważniejszej w danych warunkach). Zapisywanie
glukozy we wszystkich omawianych reakcjach wyłącznie wzorem C6H12O6 trudno nazwać nauczaniem chemii - raczej jest to publiczna egzekucja tej pięknej nauki.
A dla uczących się uwaga - żeby zbyt szybko nie rozgrzeszali swojego intelektualnego lenistwa argumentem kiepskiego
nauczyciela - pamiętajcie, że liczy się tylko WASZA rzetelna wiedza na dany temat. Jeśli trafił wam się kiepski nauczyciel, to wasz problem i kłopot, ale w niczym to was nie rozgrzesza. Dobry chemik zna dobrze chemię i w momencie
kiedy musimy się tą wiedzą wykazać, poziom naszych nauczycieli nie ma nic do rzeczy. Nauczyciel często nie wspomina o zawiłościach chemii, bo go nikt o to nie pyta. Zanim wydamy opinię o naszym mistrzu, sprawdźmy najpierw jaki
on rzeczywiście jest. Czyli najpierw sami zacznijmy zastanawiać się nad problemami poruszanymi na wykładzie czy
przeczytanymi w podręczniku, a później wdajmy się w dyskusję z nauczycielem. Raz jeszcze powtórzmy sobie niepodważalną prawdę - chemii nie da się nauczyć na pamięć, bez zrozumienia jej zasad, reguł i wzajemnych relacji między nimi. Dopiero widzenie zjawiska w kontekście całej chemii i fizyki pozwala zrozumieć jego przebieg i jego istotę.
Jak wiemy, w przyrodzie występuje spontaniczne dążenie energii i materii do maksymalnego rozproszenia. Ramy zakreślające zakres tych zjawisk wyznaczają warunki, w jakich znajduje się dany układ, oraz opozycyjność tych zjawisk.
Rozproszenie (zmiana stopnia uporządkowania) materii najczęściej związana jest ze wzrostem energii, a obniżanie
energii często prowadzi poprzez uporządkowania materii (np. łączenie się atomów w cząsteczki).
Czasami jednak trudno zrozumieć, czemu np. jony w roztworze nie łączą się w niezdysocjowane cząsteczki, skoro
układ odpychających się jednoimiennych jonów i przyciągających się jonów różnoimiennych jest bogaty energetycznie.
Dodatkowe wątpliwości powstaną, gdy zaczniemy się zastanawiać, dlaczego jony Ag+ i Cl- momentalnie w roztworze
łącza się w niezdysocjowane AgCl i dodatkowo wypadają w postaci nierozpuszczalnego osadu, a jony Na + i Cl- istnieją obok siebie w roztworze bez problemów. Jeszcze inne związki rozpuszczają się dobrze ale dysocjują tylko częściowo
(słabe elektrolity, np. kwas octowy). Jeszcze bardziej skomplikowane i trudne do wyjaśnienia jest zjawisko wysycania
roztworu wodorotlenku wapnia ditlenkiem węgla. Najpierw wypada osad węglanu wapnia, później osad rozpuszcza się
przechodząc w wodorowęglan wapnia, by po podgrzaniu (dostarczeniu energii!) wypaść znów w postaci osadu węglanu.
Przebiegu wielu zjawisk nie rozumiemy najczęściej tak długo, jak długo nie włączymy do rozważań innych czynników
biorących udział w zjawisku. Najczęściej zapominamy o ... rozpuszczalniku, oraz innych (prócz rozpatrywanego) składnikach analizowanego układu (roztworu). Jesteśmy też skłonni uważać stan poszczególnych elementów układu za zgodny z symbolicznym zapisem - zapominamy o wiązaniach wodorowych między cząsteczkami substancji rozpuszczonej,
między cząsteczkami rozpuszczalnika, oddziaływaniem substancji rozpuszczonej (lub jonu) z cząsteczkami rozpuszczalnika (solwatacja), itd. Jeżeli te "niezauważone" czynniki wnoszą w konkretnym przypadku istotny wkład do wartości entalpii swobodnej układu, ich pominięcie w rozważaniach może prowadzić do niezrozumienia zachowania się badanego układu.
Jeżeli rozpuszczamy w wodzie tiosiarczan sodu bezwodny (Na2S2O3), to roztwór bardzo silnie się rozgrzewa. W tym
przypadku egzoenergetyczny proces solwatacji powoduje wydzielenie takiej ilości energii na sposób ciepła, że znacznie
przewyższa ona ilość energii pochłoniętej w procesie rozpuszczania. Energia pochłaniana jest przez sieć krystaliczną, w
celu uwolnienia cząsteczek, oraz przez zjawisko dysocjacji, a właściwie hydrolizy tiosiarczanu. Jeśli jednak rozpuszczać będziemy tiosiarczan pięciowodny (Na2S2O3*5H2O) to roztwór bardzo silnie się ochłodzi. W tym przypadku
proces hydratacji nie wystąpi (lub wystąpi w stopniu znacznie mniejszym), bowiem cząsteczki tiosiarczanu już są
uwodnione. O wielkości efektu zadecyduje proces zrywania wiązań sieci krystalicznej i wiązań jonowych (endoenergetyczny) i cały proces będzie endotermiczny.
Ustalając, co w danym przypadku wziąć pod uwagę, a co chwilowo odrzucić jako mało istotne (wedle zasady "nie
wszystko na raz") nie zapominajmy nigdy rozważyć wpływu oddziaływań międzycząsteczkowych, zarówno substancji
jak i środowiska (rozpuszczalnika) oraz, często nieuświadomionego, procesu solwatacji polarnych cząsteczek czy jonów w polarnym rozpuszczalniku. Nie zapominajmy też o drugim czynniku "dzielącym", utrzymującym na dystans elementy mające naturalne skłonności do łączenia się (jony przeciwnych znaków!) - o energii translacji cząsteczek (temperatura). Jeśli tytuł tego rozdziału potraktujemy jako pytanie, to odpowiedź będzie brzmiała - przede wszystkim otoczki
solwatacyjne i ruch translacyjny. W niektórych przypadkach (koloidy, emulsje) także ładunek na powierzchni cząstek.