Padaczka i encefalopatie padaczkowe
Padaczki i encefalopatie padaczkowe to szeroka grupa zaburzeń, wśród których 80% ma
podłoże genetyczne. W oparciu o rodzaje napadów, padaczki można podzielić na uogólnione,
ogniskowe oraz encefalopatie padaczkowe.
Postępujące encefalopatie mogą prowadzić do stopniowej utraty funkcji ruchowych
i poznawczych, współistniejącej z pogłębiającymi się zaburzeniami neurologicznymi.
W niniejszym teście, dzięki nowoczesnej technologii sekwencjonowania genomowego,
badamy pełne sekwencje 194 genów, odpowiedzialnych za padaczkę i encefalopatie
padaczkowe.
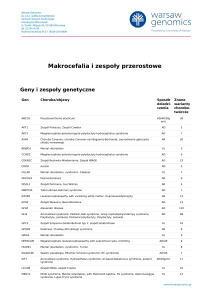
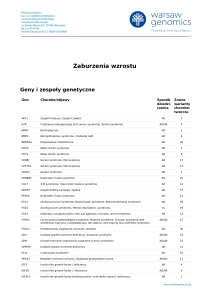
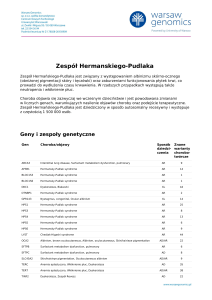
Geny i zespoły genetyczne
Gen
Choroba/objawy
ABCD1
Adrenoleukodystrofia; Choroba Addisona i stwardnienie guzowate; choroba SiemerlingaCreutzfeldta; choroba Schildera; Melanodermia
ADAR
Dyschromatosis symmetrica hereditaria, Aicardi-Goutières syndrome
ADSL
Sposób Znane
dziedzi- warianty
czenia
chorobotwórcze
XL
55
AD/AR
17
Deficyt liazy adenylobursztynianowej
AR
22
AFG3L2
Spastic ataxia, Spinocerebellar ataxia
AD/AR
20
AGA
Aspartylglucosaminuria
AR
28
AIMP1
Leukodystrophy, hypomyelinating
AR
2
ALDH5A1
Succinic semialdehyde dehydrogenase deficiency
AR
6
ALDH7A1
Padaczka pirydoksyno-zależna
AR
23
ALG13
Congenital disorder of glycosylation
XL
3
AMACR
Alpha-methylacyl-CoA racemase deficiency, Bile acid synthesis defect
AR
3
AMT
Glycine encephalopathy
AR
26
ARG1
Hyperargininemia
AR
13
ARHGEF9
Wczesna encefalopatia padaczkowa niemowląt, typ 8
XL
2
ARSA
Metachromatic leukodystrophy
AR
63
ARX
Wczesna encefalopatia padaczkowa niemowląt, typ 1, Zespół Westa
XL
54
ASAH1
Spinal muscular atrophy with progressive myoclonic epilepsy, Farber lipogranulomatosis
AR
12
Gen
Choroba/objawy
Sposób Znane
dziedzi- warianty
czenia
chorobotwórcze
ASPA
Choroba Canavan; choroba Canavan-van Bogaerta-Bertranda; zwyrodnienie gąbczaste
układu nerwowego
AR
20
ATP13A2
Parkinson disease (Kufor-Rakeb syndrome)
AR
11
ATRX
Alfa talasemia z upośledzeniem umysłowym
XL
38
BTD
Biotinidase deficiency
AR
168
CACNA1A
Migraine, familial hemiplegic, Episodic ataxia
AD
45
CACNA1H
Childhood absence epilepsy
AD
3
CACNB4
Padaczka idiopatyczna, podatność, typ 9
AD
1
CASK
Mental retardation and microcephaly with pontine and cerebellar hypoplasia, FG
syndrome, Mental retardation
XL
33
CASR
Nadczynność przytarczyc, Hipokalcemia
AD/AR
74
CDKL5
Wczesna encefalopatia padaczkowa niemowląt, typ 2
XL
207
CERS1
Epilepsy, progressive myoclonic
AR
4
CHD2
Encefalopatia padaczkowa dziecięca
AD
33
CHRNA2
Epilepsy, nocturnal frontal lobe
AD
1
CHRNA4
Epilepsy, nocturnal frontal lobe
AD
7
CHRNB2
Epilepsy, nocturnal frontal lobe
AD
8
CLCN2
Młodzieńcza padaczka miokloniczna, podatność
AD/AR
18
CLN3
Ceroidolipofuscynoza neuronalna typ 3; choroba Battena, choroba Vogta-Spielmeyera,
choroba Spielmeyera-Sjögrena
AR
64
CLN5
Ceroidolipofuscynoza neuronalna typ 5
AR
40
CLN6
Ceroidolipofuscynoza neuronalna typ 6
AR
22
CLN8
Ceroidolipofuscynoza neuronalna typ 8
AR
31
CNTNAP2
Dysplazja korowa - zespół ogniskowej padaczaki; zespół CDFE
AR
20
COL4A1
Schizencephaly, Anterior segment dysgenesis with cerebral involvement, Retinal artery
tortuosity, Porencephaly, Angiopathy, hereditary, with nephropathy, aneurysms, and
muscle cramps, Brain small vessel disease
AD
25
COX15
Leigh syndrome, Cardioencephalomyopathy, fatal infantile, due to cytochrome c oxidase
deficiency
AR
7
CPT2
Carnitine palmitoyltransferase II deficiency
AR
37
CSF1R
Leukoencephalopathy, diffuse hereditary, with spheroids
AD
44
CSTB
Padaczka miokloniczna Unverrichta-Lundborga
AR
16
CTSD
Ceroidolipofuscynoza neuronalna typ 10
AR
11
CTSF
Ceroidolipofuscynoza neuronalna typ 13
AR
6
CUL4B
Mental retardation, syndromic, Cabezas
XL
7
DARS2
Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation
AR
13
DCX
Lissencephaly, Subcortical laminal heterotopia
XL
114
DEPDC5
Epilepsy, familial focal, with variable foci
AD
15
DNAJC5
Kufs disease,, Ceroid lipofuscinosis, neuronal 4, Parry
AD
2
DNM1
Epileptic encephalopathy, early infantile
AD
7
Gen
Choroba/objawy
DOCK7
Epilepitic encephalopathy
DPYD
analogi pirymidyn
EARS2
Sposób Znane
dziedzi- warianty
czenia
chorobotwórcze
AR
11
AD/AR
3
Combined oxidative phosphorylation deficiency
AR
7
EEF1A2
Epileptic encephalopathy, early infantile, Mental retardation
AD
3
EFHC1
Młodzieńcza padaczka miokloniczna; zespół Janza, podatność
AD/AR
3
EIF2B1
Leukoencephalopathy with vanishing white matter, Ovarioleukodystrophy
AR
8
EIF2B2
Leukoencephalopathy with vanishing white matter, Ovarioleukodystrophy
AR
7
EIF2B3
Leukoencephalopathy with vanishing white matter, Ovarioleukodystrophy
AR
5
EIF2B4
Leukoencephalopathy with vanishing white matter, Ovarioleukodystrophy
AR
6
EIF2B5
Leukoencephalopathy with vanishing white matter, Ovarioleukodystrophy
AR
13
EPM2A
Postępująca padaczka miokloniczna typ 2B, zespół Lafora
AR
12
ETFA
Glutaric aciduria, Multiple acyl-CoA dehydrogenase deficiency
AR
6
ETFB
Glutaric aciduria, Multiple acyl-CoA dehydrogenase deficiency
AR
7
ETFDH
Glutaric aciduria, Multiple acyl-CoA dehydrogenase deficiency
AR
32
FAM126A
Leukodystrophy, hypomyelinating
AR
5
FH
Wrodzona mięśniakowatość gładkokomórkowa (leiomiomatoza) i rak nerkowokomórkowy
AD
81
FLNA
Intestinal pseudoobstruction, neuronal/Congenital short bowel syndrome, Heterotopia,
periventricular, Ehlers-Danlos variant, Cardiac valvular dysplasia
XL
78
FOLR1
Choroba neurodegeneracyjna związana z nieprawidłowym transportem folianów do mózgu
AR
5
FOXG1
Zespół Retta
AD
61
FOXRED1
Leigh syndrome, Mitochondrial complex I deficiency
AR
10
GABRA1
Młodzieńcza padaczka miokloniczna, podatność
AD
11
GABRB3
Padaczka wieku dziecięcego, brak, podatność
AD
1
GABRG2
Zespół Dravet związany z mutacjami w genie GABRG2
AD
20
GALC
Choroba Krabbego; leukodystrofia globoidalna
AR
29
GAMT
Zespół niedoboru mózgowej kreatyny
AR
11
GCDH
Glutaric aciduria
AR
34
GCH1
Dystonia wrażliwa na dopaminę; zespół Segawy
AD/AR
22
GFAP
Alexander disease
AD
110
GJC2
Spastic paraplegia, Lymphedema, hereditary, Leukodystrophy, hypomyelinating
AD/AR
13
GLDC
Glycine encephalopathy
AR
87
GNAO1
Epileptic encephalopathy, early infantile
AD
7
GNE
Inclusion body myopathy, Nonaka myopathy, Sialuria
AD/AR
22
GOSR2
Postępująca padaczka miokloniczna typ 6
AR
4
GPHN
Hyperekplexia, Molybdenum cofactor deficiency
AD/AR
3
GRIA3
Mental retardation
XL
9
GRIN2A
Padaczka z zaburzeniami mowy i lub bez niepełnosprawności intelektualnej
AD
29
Gen
Choroba/objawy
GRIN2B
Wczesna encefalopatia padaczkowa niemowląt typ 27
GRN
Frontotemporal lobar degeneration with TDP43 inclusions, GRN-related, Neuronal ceroid
lipofuscinosis
HCN1
Epileptic encephalopathy, early infantile
HEPACAM
Megalencephalic leukoencephalopathy with subcortical cysts, remitting
HNRNPU
Sposób Znane
dziedzi- warianty
czenia
chorobotwórcze
AD
26
AD/AR
24
AD
7
AD/AR
9
Intellectual disability and seizures
AD
5
HSD17B10
17-beta-hydroxysteroid dehydrogenase X deficiency, Mental retardation, syndromic
XL
6
HSPD1
Spastic paraplegia, Leukodystrophy, hypomyelinating
AD/AR
4
IQSEC2
Mental retardation
XL
10
KCNA1
Zespół ataksja/miokimia
AD
20
KCNA2
Epileptic encephalopathy, early infantile
AD
4
KCNB1
Early infantile epileptic encephalopathy
AD
3
KCNC1
Epilepsy, progressive myoclonic
AD
1
KCNQ2
Wczesna encefalopatia padaczkowa niemowląt typ 7
AD
147
KCNQ3
Seizures, benign neonatal
AD
13
KCNT1
Wczesna encefalopatia padaczkowa niemowląt typ 14
AD
14
KCTD7
Postępująca padaczka miokloniczna typ 3, zespół Lafora
AR
11
KDM5C
Mental retardation, syndromic, Claes-Jensen
XL
14
KIF1A
Spastic paraplegia, Neuropathy, hereditary sensory, Mental retardation
AD/AR
24
L2HGDH
L-2-hydroxyglutaric aciduria
AR
8
LGI1
Epilepsy, familial temporal lobe
AD
18
MARS2
Combined oxidative phosphorylation deficiency
AR
7
MBD5
Niepełnosprawność intelektualna dziedziczona autosomalnie dominująco
AD
15
MECP2
Zespół Angelman-like
XL
425
MED12
Ohdo syndrome, Mental retardation, with Marfanoid habitus, FG syndrome, Opitz-Kaveggia
syndrome, Lujan-Fryns syndrome
XL
12
MEF2C
Niepełnosprawność intelektualna dziedziczona autosomalnie dominująco
AD
22
MFSD8
Ceroidolipofuscynoza neuronalna typ 7
AR
18
MLC1
Megalencephalic leukoencephalopathy with subcortical cysts
AR
17
MOCS1
Molybdenum cofactor deficiency
AR
6
MTHFR
Homocystynuria związana z mutacjami w genie MTHFR
AD/AR
1
MTOR
Smith-Kingsmore syndrome
AD
6
NDUFAF5
Mitochondrial complex I deficiency
AR
5
NECAP1
Epileptic encephalopathy, early infantile
AR
1
NEU1
Sialidosis
AR
18
NHLRC1
Postępująca padaczka miokloniczna typ 2B, zespół Lafora
AR
13
NOTCH3
Miofibromatoza noworodków
AD
25
NRXN1
Zespół Pitta-Hopkinsa-like 2
AD/AR
45
Gen
Choroba/objawy
Sposób Znane
dziedzi- warianty
czenia
chorobotwórcze
OFD1
Simpson-Golabi-Behmel syndrome, Retinitis pigmentosa, Orofaciodigital syndrome,
Joubert syndrome
XL
122
OPHN1
Mental retardation, with cerebellar hypoplasia and distinctive facial appearance
XL
12
PCDH19
Wczesna encefalopatia padaczkowa niemowląt typ 9; Zespół Juberga-Hellmana
XL
57
PGK1
Phosphoglycerate kinase 1 deficiency
XL
14
PHF6
Zespół Borjesona-Forssmana-Lehmana
XL
15
PIGA
Multiple congenital anomalies-hypotonia-seizures syndrome
XL
18
PLCB1
Wczesna encefalopatia padaczkowa niemowląt typ 12
AR
5
PLP1
Choroba Pelizaeusa-Merzbachera, leukodystrofia
XL
32
PNKP
Małogłowie, drgawki i opóźnienie rozwoju
AR
23
PNPO
Niedobór oksydazy 5'-fosforanu pirydoksaminy
AR
15
POLR3A
Leukodystrophy, hypomyelinating
AR
18
POLR3B
Leukodystrophy, hypomyelinating
AR
11
PPT1
Ceroidolipofuscynoza neuronalna typ 1
AR
66
PRICKLE1
Epilepsy, progressive myoclonic
AD/AR
3
PRICKLE2
Epilepsy, progessive myoclonic
AD
1
PRODH
Hyperprolinemia
AR
16
PRRT2
Drgawki dziecięce z napadową choreoatetozą
AD
25
PSAP
Krabbe disease, atypical, Metachromatic leukodystrophy due to saposin-b deficiency,
Combined saposin deficiency, Gaucher disease, atypical, due to saposin C deficiency
AR
15
PTS
Hyperphenylalaninemia, BH4-deficient
AR
11
PURA
Mental retardation
AD
28
QDPR
Hyperphenylalaninemia, BH4-deficient
AR
8
RAB39B
Waisman parkinsonism-mental retardation syndrome, Waisman parkinsonism-mental
retardation syndrome, Mental retardation
XL
4
RELN
Lissencephaly, Epilepsy, familial temporal lobe
AD/AR
16
RNASEH2A
Aicardi-Goutières syndrome
AR
12
RNASEH2B
Aicardi-Goutières syndrome
AR
4
RNASEH2C
Aicardi-Goutières syndrome
AR
4
RNASET2
Leukoencephalopathy, cystic, without megalencephaly
AR
6
SAMHD1
Aicardi-Goutières syndrome
AR
23
SCARB2
Postępująca padaczka miokloniczna typ 4
AR
9
SCN1A
Wczesna encefalopatia padaczkowa niemowląt typ 6; Zespół Draveta
AD
444
SCN1B
Zespół Brugadów, Padaczka uogólniona z napadami gorączkowymi typu 1
AD
12
SCN2A
Wczesna encefalopatia padaczkowa niemowląt typ 11
AD
84
SCN8A
Wczesna encefalopatia padaczkowa niemowląt typ 13
AD
28
SCN9A
Zespół Dravet (modifier)
AD/AR
29
SERPINI1
Encephalopathy, familial, with neuroserpin inclusion bodies
AD
5
Gen
Choroba/objawy
Sposób Znane
dziedzi- warianty
czenia
chorobotwórcze
SIK1
Epileptic encephalopathy, early infantile
AD
5
SLC12A5
Epileptic encephalopathy, early infantile
AR
3
SLC13A5
Wczesna encefalopatia padaczkowa niemowląt typ 25
AR
9
SLC19A3
Zespół nieprawidłowego metabolizmu tiaminy
AR
11
SLC25A15
Hyperornithinemia-hyperammonemia-homocitrullinemia syndrome
AR
18
SLC25A22
Wczesna encefalopatia padaczkowa niemowląt typ 3
AR
6
SLC2A1
syndrom deficytu GLUT1
AD/AR
59
SLC35A2
Congenital disorder of glycosylation
XL
7
SLC46A1
Folate malabsorption
AR
17
SLC6A1
Myoclonic-astastic epilepsy
AD
5
SLC6A8
Creatine deficiency syndrome
XL
14
SLC9A6
Niepełnosprawność intelektualna, sprzężona z chromosomem X
XL
19
SMS
Mental retardation, Snyder-Robinson
XL
9
SNAP25
Myasthenic syndrome, congenital
AD
2
SOX10
Peripheral demyelinating neuropathy, central dysmyelination, Waardenburg syndrome,
and Hirschsprung disease
AD
28
SPTAN1
Wczesna encefalopatia padaczkowa niemowląt typ 5
AD
16
ST3GAL3
Wczesna encefalopatia padaczkowa niemowląt typ 15
AR
3
ST3GAL5
Ganglioside GM3 synthase deficiency
AR
2
STX1B
Generalized epilepsy with febrile seizures plus
AD
5
STXBP1
Wczesna encefalopatia padaczkowa niemowląt typ 4
AD
52
SUMF1
Multiple sulfatase deficiency
AR
18
SUOX
Sulfocysteinuria
AR
6
SYN1
Epilepsy, with variable learning disabilities and behavior disorders
XL
6
SYNGAP1
Mental retardation
AD
20
SZT2
Wczesna encefalopatia padaczkowa niemowląt typ 18
AR
3
TBC1D24
Wczesna encefalopatia padaczkowa niemowląt typ 16
AD/AR
26
TCF4
Zespół Pitta-Hopkinsa; encefalopatia; ciężkie napady padaczkowe z dysfunkcją
autonomicznego układu nerwowego
AD
46
TPP1
Ceroidolipofuscynoza neuronalna typ 2
AR
28
TREX1
Vasculopathy, retinal, with cerebral leukodystrophy, Chilblain lupus, Aicardi-Goutières
syndrome
AD/AR
22
TSC1
Stwardnienie guzowate, Limfangioleiomiomatoza
AD
43
TSC2
Stwardnienie guzowate, Limfangioleiomiomatoza
AD
99
TUBB4A
Leukodystrophy, hypomyelinating, Dystonia
AD
11
UBE2A
Mental retardation, syndromic, Nascimento
XL
3
UBE3A
Zespół Angelmana
AD
133
WDR45
Neurodegeneration with brain iron accumulation
XL
12
Gen
Choroba/objawy
Sposób Znane
dziedzi- warianty
czenia
chorobotwórcze
WWOX
Wczesna encefalopatia padaczkowa niemowląt typ 28
AR
17
ZEB2
Zespół Mowata-Wilsona
AD
94
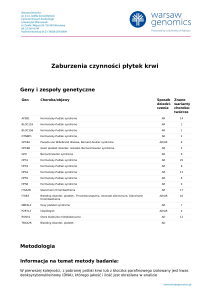
Metodologia
Informacja na temat metody badania:
W pierwszej kolejności, z pobranej próbki krwi lub z bloczka parafinowego izolowany jest kwas
deoksyrybonukleinowy (DNA), którego jakość i ilość jest określana w analizie
spektrofotometrycznej i fluorymetrycznej. Po mechanicznej lub enzymatycznej fragmentacji,
DNA jest wykorzystywany do stworzenia biblioteki, umożliwiającej oznaczenie, a następnie
zsekwencjonowanie i analizę genów, które zostały wybrane w ramach zleconego panelu.
Otrzymana biblioteka jest sekwencjonowana na sekwenatorze nowej generacji. Otrzymane
wyniki zostają następnie poddane analizie bioinformatycznej i interpretacji klinicznej. Warianty
genetyczne są identyfikowane z wykorzystaniem Burrows-Wheeler Aligner. Test umożliwia
wykrycie 100% substytucji i 95% małych insercji i delecji.
Informacja na temat klasyfikacji wariantów:
W raporcie z badania przedstawiana jest informacja na temat wariantów zaklasyfikowanych
jako warianty „potencjalnie patogenne” i „patogenne”, z uwagi na ich potencjalne znaczenie
kliniczne. Zidentyfikowane warianty są klasyfikowane do następujących kategorii:
Wariant patogenny: znaleziona zmiana w sekwencji genu ma bezpośredni związek
z powstawaniem choroby. Równocześnie, niektóre zmiany patogenne mogą nie mieć pełnej
penetracji, tj. pojedyncza zmiana może być niewystarczająca do wywołania pełnoobjawowej
choroby.
Wariant potencjalnie patogenny: znaleziona zmiana w sekwencji genu jest z dużym
prawdopodobieństwem związana z powstawaniem choroby, jednakże udowodnienie tego
związku nie jest możliwe w oparciu o aktualnie dostępne dane naukowe. Potwierdzenie
patogenności wariantu wymaga dodatkowych badań i dowodów; nie można wykluczyć, że
dalsze badania wykażą, że znaleziona zmiana ma niewielkie lub żadne znaczenie kliniczne.
Wariant o nieznanej patogenności: w oparciu o aktualnie dostępne dane naukowe nie ma
możliwości określenia znaczenia znalezionej zmiany.
Wariant potencjalnie łagodny: znaleziona zmiana w sekwencji genu najprawdopodobniej nie
ma związku z powstawaniem choroby, jednakże w oparciu o aktualnie dostępne dane naukowe
nie ma możliwości potwierdzenia łagodności zmiany. Potwierdzenie klinicznego znaczenia
wariantu wymaga dodatkowych badań i dowodów; nie można wykluczyć, że dalsze badania
wykażą, że znaleziona zmiana ma znaczenie kliniczne i prowadzi do rozwinięcia choroby
Wariant łagodny: znaleziona zmiana nie ma związku z powstawaniem choroby
Zidentyfikowane warianty genetyczne klasyfikowane są w oparciu o wytyczne opracowane
przez American College of Medical Genetics and Genomics i American Association for Molecular
Pathology (S. Richards, Genet Med. 2015 May;17(5):405-24). W klasyfikacji wariantów brane są
pod uwagę następujące kryteria:
wcześniejsza identyfikacja wariantu u osób obciążonych chorobą
wpływ wariantu na powstawanie funkcjonalnego produktu genu:
określony w analizach bioinformatycznych
potwierdzony w badaniach in vitro/in vivo
lokalizacja wariantu (ekson/intron, domena funkcjonalna)
zmiana de novo/dziedziczna
częstość występowania wariantu w populacji ogólnej (każdy wariant występujący
z częstością >5% zgodnie z Exome Sequencing Project, 1000 Genomes Project lub
Exome Aggregation Consortium jest klasyfikowany jako zmiana łagodna)
częstość występowania wariantu w populacji ogólnej w stosunku do populacji osób
chorych
Ostateczna klasyfikacja wariantów prowadzona jest w oparciu o sumę wymienionych kryteriów.
Przeszukiwane bazy danych obejmują: 1000GP, ClinVar, ConsensusPathDB, Exome Aggregation
Consortium, Exome Variant Server, FATHMM, GO (Gene Ontology), GTEx (Genotype-Tissue
Expression), GWAS (Genome Wide Association Study), HGMD, KEGG, MetaLR, MetaSVM,
MutationAssessor, MutationTaster, OMIM, PolyPhen-2, PROVEAN, SIFT, SnpEff, dbNSFP, UniProt,
VEP (Variant Effect Predictor).
Ograniczenia badania:
Wszystkie technologie sekwencjonowania mają swoje ograniczenia. Zlecane badanie jest
wykonywane z wykorzystaniem sekwencjonowania nowej generacji (NGS) i ma na celu
zbadanie regionów kodujących i splicingowych zleconych genów. Chociaż stosowane techniki
sekwencjonowania oraz późniejsze analizy bioinformatyczne są ukierunkowane na ograniczenie
znaczenia sekwencji pseudogenów, to jednak obecność wysoce homologicznych sekwencji
genowych może nadal sporadycznie zakłócać zdolność identyfikacji patogennych alleli, jak
i delecji/duplikacji. Sekwencjonowanie Sangera jest metodą wykorzystywaną do potwierdzania
wariantów, które uzyskały niższe parametry jakości. Analizy delecji/duplikacji wskazują na
zmiany ilościowe DNA obejmujące minimum jeden ekson i zawsze wymagają potwierdzenia
innymi metodami (qPCR lub MLPA). Wykonane analizy nie są przeznaczone do wykrywania
pewnych typów zmian genomowych, jak translokacje, inwersje, mutacje dynamiczne (np.
zwiększenie ilości powtórzeń trzynukleotydowych), zmian w regionach regulatorowych czy
intronowych. Jeśli raportowane jest zwiększenie liczby powtórzeń dwu- czy trzynukleotydowych,
to trzeba założyć, że dokładna liczba powtórzeń nie jest precyzyjna. Przeprowadzane badanie
nie jest przeznaczone do wykrywania mozaikowatości somatycznych, a analizy mutacji
somatycznych powinny być prowadzone w kontekście sekwencji DNA germinalnego.
Nie ma możliwości wykluczenia obecności mutacji w genach i rejonach innych niż objęte
wykonywanym badaniem, a także zmian liczby kopii genu. Raport z badania zawiera informację
na temat zmian w sekwencji genów zidentyfikowanych w oparciu o porównanie z aktualnymi
sekwencjami referencyjnymi zdeponowanymi w bazach danych NCBI Nucleotide i Ensembl.
Testy są opracowywane w Warsaw Genomics do celów klinicznych. Wszystkie otrzymywane
wyniki badań są interpretowane i analizowane przez ekspertów naukowych i medycznych
Warsaw Genomics.
Jak zlecić badanie
Informacja na temat metody badania:
Badanie można zlecić bezpośrednio na stronie internetowej Warsaw Genomics, poprzez
zaznaczenie wybranego testu. Zalecamy jednak, by przed każdym badaniem skonsultować się
z lekarzem, który pomoże w wybraniu odpowiedniego testu diagnostycznego, wyjaśni
możliwości i ograniczenia testów genetycznych a także przedstawi możliwe wyniki
i konsekwencje przeprowadzenia badania.
Czas realizacji badania: od 4 do 10 tygodni. Będziemy informować o kolejnych etapach analizy.
KONSULTACJA LEKARSKA
REJESTRACJA
Wybranie właściwego testu
genetycznego lub
indywidualnego zestawu
genów
Wypełnienie formularza
zlecenia testu.
Formularz wypełnia pacjent
lub wybrany przez niego
lekarz.
OPŁACENIE TESTU
Po wykonaniu testu
POBRANIE KRWI
Łącznie należy pobrać 4ml
krwi do jednej probówki
z EDTA (takiej jak na
morfologie). Pobraną krew
można przechowywać
w lodówce (w temp. +4st C)
do 7 dni
WYNIK BADANIA
PRZESŁANIE PRÓBKI
KRWI NA NASZ ADRES
Próbkę można dostarczyć
osobiście lub kurierem
(w temperaturze pokojowej)
w ciągu 48 godzin.
Szczegółowa instrukcja
pakowania próbki
i zamówienia kuriera jest
dostępna tutaj. Do próbki
dołączamy wydrukowany
i podpisany formularz
zlecenia testu.
KONSULTACJA LEKARSKA
zostanie przekazany osobie
zlecającej test – pacjentowi
lub wybranemu przez niego
lekarzowi
Jak przekazać materiał do badania?
Badanie genetyczne z krwi:
1. Krew należy pobrać do jednej probówki z EDTA (nie pobierać do probówek na skrzep,
ani na heparynę litową). Pobranie krwi może nastąpić w dowolnej godzinie, pacjent nie
musi być na czczo:
osoba dorosła - ok. 4 ml krwi żylnej do izolacji DNA (pobraną krew należy
dokładnie wymieszać z antykoagulantem i przechowywać w temperaturze 4°C)
dzieci – ok. 4 ml (minimalna ilość 2 ml) krwi żylnej do izolacji DNA (pobraną
krew należy dokładnie wymieszać z antykoagulantem i przechowywać
w temperaturze 4°C)
niemowlę – ok. 1,5 - 2 ml krwi żylnej do izolacji DNA (pobraną krew należy
dokładnie wymieszać z antykoagulantem i przechowywać w temperaturze 4°C)
2. Probówkę należy opisać imieniem i nazwiskiem i zabezpieczyć (można zakleić taśmą
klejącą).
3. Zabezpieczoną probówkę wraz z wypełnionym i podpisanym formularzem zlecenia
badania należy zapakować i wysłać zgodnie z instrukcją dostępną pod adresem:
https://badamygeny.pl/BADAMY_GENY/docs/instrukcja-wysylki-probki-krwi.pdf
Badanie genetyczne z bloczka parafinowego (profilowanie
nowotworu):
1. Należy pobrać łącznie 4 ml krwi do jednej probówki z EDTA (nie pobierać do probówek
na skrzep, ani na heparynę litową). Pobranie krwi może nastąpić w dowolnej godzinie,
pacjent nie musi być na czczo. Pobraną krew należy dokładnie wymieszać
z antykoagulantem i przechowywać w temperaturze 4°C.
2. Probówkę należy opisać imieniem i nazwiskiem i zabezpieczyć (można zakleić taśmą
klejącą).
3. Uzyskanie tkanki nowotworowej do badania w postaci:
bloczka parafinowego zawierającego wycinek nowotworu wraz z uzyskanym
z bloczka preparatem histopatologicznym (szkiełkiem) umożliwiającym
zlokalizowanie fragmentu tkanki nowotworowej,
albo wycinka tkanki nowotworowej z bloczka parafinowego o wymiarach min.
4x4x1mm, zawierającego wyłącznie tkankę nowotworową.
4. Próbkę należy opisać imieniem i nazwiskiem i zabezpieczyć (można zakleić taśmą
klejącą).
5. Zabezpieczony materiał wraz z wypełnionym i podpisanym formularzem zlecenia
badania należy zapakować i wysłać zgodnie z instrukcją dostępną pod adresem:
https://badamygeny.pl/BADAMY_GENY/docs/instrukcja-wysylki-probki-krwi.pdf
Powered by TCPDF (www.tcpdf.org)